质粒转染的操作步骤有哪些?
6 个回答

关注公众号“医学科研小坑”,了解更多相关科研技术内容!
概念:概括地说,转染是使用非病毒感染的手段人工引入核酸(DNA或RNA)进入细胞的过程。转染的目的是产生重组蛋白,或特异性增强或抑制转染细胞中的基因表达。
转染的类型
根据导入的核酸存在于宿主细胞的时间长短,可以分为瞬转和稳转。根据转染方式可以分为化学,生物,物理方法。
瞬转:因为导入的核酸没有整合到宿主细胞基因组,因此它只会短暂地存在于宿主细胞中,不会随着细胞的分裂而进入到子代细胞中。然而,导入的遗传物质的高拷贝数导致其在细胞内的蛋白质表达水平较高。根据所使用的载体的不同,瞬转通常可以1至7天内进行基因检测,但是瞬时转染的细胞通常在转染后24-96小时收获。当使用超螺旋质粒DNA时,瞬时转染的效果最好,推测是由于超螺旋质粒DNA能更有效地被细胞摄取。
稳转:外源DNA整合到细胞基因组中或作为附加体质粒保留在细胞中。与瞬时转染不同,稳定转染允许外源DNA在转染的细胞及其后代中的长期维持。然而,通常是将单拷贝或几个拷贝的外源DNA整合到稳定转染的细胞的基因组中,因此,其表达水平一般低于瞬时转染的表达水平。
由于稳转效率较低,因此选用有效地转染策略和筛选方法很重要。其中比较可靠地筛选方法是在DNA载体中包含选择性标记,然后在细胞转染短暂性恢复后进行适当地选择性加压。尽管相对于超螺旋DNA,线性DNA被细胞摄取的效率较低,但其能最有效地整合到宿主基因组中。
目前,由于各种转染试剂的发明,哺乳动物细胞的瞬时转染已经用于生产具有适当折叠和翻译后修饰的重组蛋白。但表达mg/L-g/L的重组蛋白主要依赖于稳定细胞系的产生。
瞬转和稳转的比较
| 瞬转 | 稳转 |
| 导入的DNA没有整合到基因组中,而是保留在细胞核上 | 导入的DNA整合到基因组中 |
| 导入的遗传物质不传递到子代;遗传改变只是暂时的 | 导入的遗传物质能够代代相传;遗传改变是永久的 |
| 不需要选择性筛选 | 需要选择性筛选出稳定转染的细胞 |
| DNA载体和RNA都可用于瞬时转染 | 只有DNA载体可用于稳定转染;RNA本身不能稳定地导入细胞中 |
| 导入的遗传物质的高拷贝数导致高水平的蛋白质表达。 | 单拷贝数或低拷贝数的稳定整合的DNA导致较低水平的蛋白质表达。 |
| 通常在转染后24-96小时内收获细胞。 | 需要2-3周的时间筛选出稳定转染的细胞克隆。 |
| 通常不适合使用具有诱导型启动子的载体的研究。 | 适用于使用带有诱导型启动子的载体的研究。 |
转染方式
细胞由带负电荷的磷脂双分子层构成,这对大分子物质来说是个不可透过的屏障,比如DNA和RNA的磷酸骨架,其也带负电荷。为了使核酸穿过细胞膜,研究人员开发了多种技术,大致分为三类:化学方法---利用载体分子包被核酸使其呈现中性电荷或正电荷。生物方法---利用基因工程病毒转染非病毒基因到细胞中。物理方法---在细胞膜表面产生一个瞬时的孔从而导入DNA。然而,没有一种方法适用于所有的细胞和实验,理想的方法应根据您的细胞类型和实验需要进行选择,理想的方法应具有高转染效率,低细胞毒性和对正常生理学的影响最小,并且易于使用和可重复性等特点。
| 化学转染方法 | ||
| 技术 | 优点 | 缺点 |
| 阳离子脂质体 | 操作快速简单 结果可重复 转染效率高可转染DNA,RNA和蛋白质 适用于生产瞬时和稳定的蛋白质 可用于体内转染 | 需进行条件优化(一些细胞系对阳离子脂质体较敏感) 有些细胞系不容易转染 血清的存在干扰复合物的形成,导致低转染效率 培养基中血清的缺失会增加细胞毒性 |
| 磷酸钙共沉淀 | 便宜且容易获得 适用于生产瞬时和稳定的蛋白质 转染效率高(不限制细胞系) | 需要仔细制备试剂 – CaPO4溶液对pH,温度和缓冲盐浓度的变化敏感可重复性较差 有细胞毒性,尤其对原代细胞 不能采用RPMI培养基,由于其含高浓度的磷酸盐 不适用于动物体内转染 |
| 葡聚糖 | 操作简单 结果可重复 便宜 | 对某些细胞有化学毒性 只限于瞬时转染 转染效率低,尤其在原代细胞中 |
| 其他阳离子聚合物 | 在血清中稳定,对温度不敏感 高转染效率(限制细胞系) 结果可重复 | 对某些细胞有毒性 不能生物降解(树枝状大分子) 只限于瞬时转染 |
| 生物转染方法 | ||
| 病毒转染 | 高转染效率(对原代细胞有80-90%) 适用于较难转染的细胞系 可用于体内转染 可用于构建稳定表达或瞬时表达的细胞系 | 被转染的细胞系必须含有病毒受体 基因插入大小受限(病毒载体~10 kb,非病毒载体~100 kb) 技术难度高,且构建重组蛋白很费时存在生物安全问题(激活潜在疾病,免疫原性反应,细胞毒性,插入突变,使细胞恶性转化) |
| 物理转染方法 | ||
| 电转 | 原理简单 条件优化后可产生重复性的结果 不需要载体 不限制细胞类型和条件 条件优化后可以快速转染大量细胞 | 需要特殊的设备 需要优化电转脉冲和电压参数 对细胞伤害很大 细胞死亡率很高因此需要大量细胞 会不可逆转地损坏细胞膜,溶解细胞 |
| 生物传递粒子传递(粒子轰击) | 不限制细胞类型和条件 可用于动物体内转染 方法直接,结果可靠 不限制导入基因的大小和数量 主要用于基因疫苗和农业应用 | 需要昂贵的设备 会对样品产生物理损伤细 胞死亡率很高因此需要大量细胞 需要准备微粒 转染效率相对较低 对于研究应用成本较昂贵 |
| 显微注射 | 不限制细胞类型和条件 可以单细胞转染 方法直接,结果可靠 不限制导入基因的大小和数量 不需要载体 | 需要昂贵的设备 有技术要求,且是劳动密集型(一次只能转染一个细胞) 常引起细胞死亡 |
| 激光介导的转染(光转染) | 可用于转染DNA,RNA,蛋白质,离子,葡聚糖,小分子和半导体纳米晶体 可用于非常小的细胞 允许单细胞转染或同时转染大量细胞不需要载体 转染效率高 适用于多种细胞系 | 需要昂贵的激光显微镜系统 需要贴壁细胞 有技术要求 |
阳离子脂质体介导的转染
原理:阳离子脂质体介导的转染是目前最常用的转染方式之一。阳离子脂质的基本结构由带正电荷的头基和一个或两个烃链组成,带正电荷的头基与带负电荷的核酸通过静电作用形成复合物,经细胞的内吞作用进入细胞。LifeTechnologies™提供了广泛的阳离子脂质介导的转染试剂,用于有效地将DNA,RNA,siRNA或寡核苷酸引入广泛的细胞类型,包括Lipofectamine®3000试剂。当选择转染试剂时,您必须考虑您希望导入(DNA,RNA或蛋白质)的有效载荷以及要转染的细胞类型,因为转染试剂的选择强烈影响转染结果。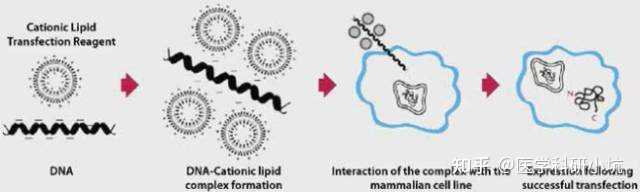
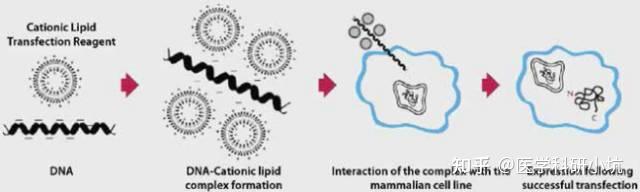
实验步骤:将DNA,RNA,siRNA或寡核苷酸和转染试剂分别在不同的管中稀释→将两者混合形成混合物→将形成的混合物加入细胞中,脂质体的正电荷有助于帮助复合物粘附到细胞膜上→复合物经细胞内吞作用进入细胞→检测基因表达或沉默情况。
磷酸钙共沉淀
原理:将DNA与氯化钙在磷酸缓冲盐水中混合形成磷酸钙-DNA沉淀物,然后将其分散在培养的细胞上,磷酸钙促进共沉淀物中的缩合DNA与细胞表面的结合,DNA通过内吞作用进入细胞。
实验步骤:将氯化钙和DNA混合,以可控的方式加入磷酸缓冲液。→在室温下孵育,以形成极细,可溶的共沉淀微粒→将磷酸钙-DNA沉淀物加入细胞中,后者能黏附到细胞膜表面→共沉淀物经细胞内吞作用进入细胞→检测基因表达或沉默情况。
病毒转染
原理:对于用脂质体不能实现转染的细胞,可以采用病毒转染。腺病毒,逆转录病毒和慢病毒载体已广泛用于哺乳动物细胞体内外的基因转染。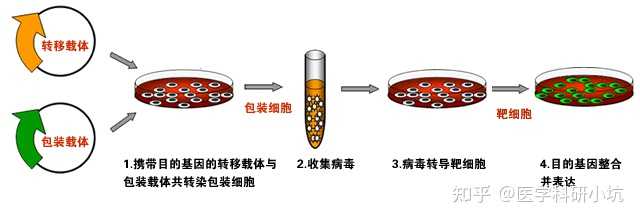
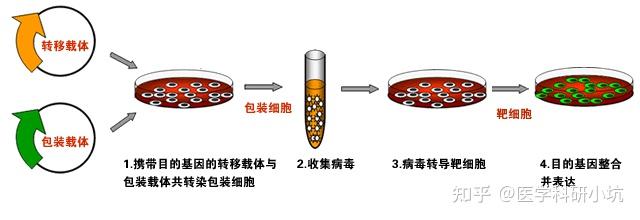
实验步骤:通过基因克隆方法获得重组病毒表达载体→转染包装细胞系,扩增并分离得到重组病毒颗粒→纯化并滴定病毒液→转导目的细胞(含有病毒特异性的受体)→从培养基中移除病毒→检测基因表达或沉默情况。
电转
原理:利用电脉冲在细胞膜上形成暂时的孔使核酸物质能穿过孔进入细胞。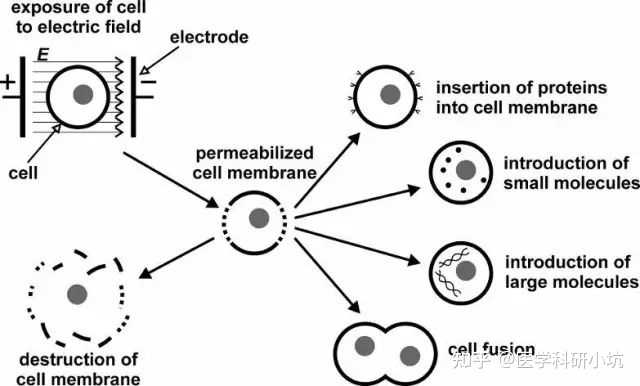
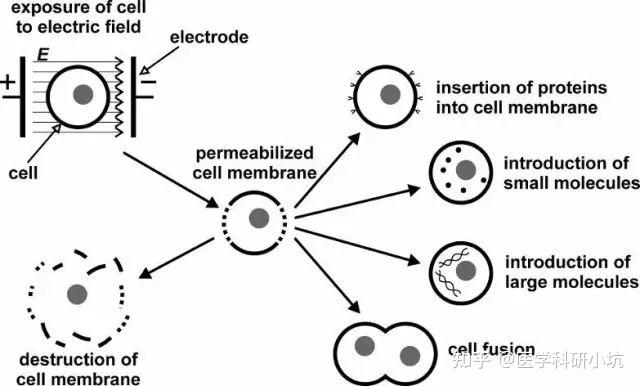
实验步骤:利用电转缓冲液重悬细胞→对含有核酸,缓冲液,细胞的混合物给予合适的电脉冲→电脉冲在细胞膜上形成电势差,诱导产生暂时的孔使核酸进入细胞→将细胞返回到生长培养基中,使其慢慢恢复→检测基因表达或沉默情况。
转染的影响因素
转染过程中许多因素会影响转染效率:如细胞种类、细胞密度;质粒大小、质粒纯度;血清;抗生素;转染试剂等。
转染实验中,以下几个因素需多加注意。
1. 转染前的细胞状态和密度
转染时细胞密度以 70~90%(贴壁细胞)或 2×10的6~4×10的6次方细胞 / mL(悬浮细胞)为宜,细胞密度过高会严重影响细胞状态(周期进程阻滞,凋亡增加等)。转染效率随着细胞传代次数不同会发生很大变化,因为传代次数少的细胞转染率非常低,而对传代次数较多的细胞而言,其转染最佳剂量差别较大。同时支原体污染会严重降低细胞的转染效率,因此转染前可用环丙沙星处理细胞以除去支原体。
2. 转染时的质粒纯度
假使质粒不纯,如带少量盐离子,蛋白、代谢物污染等都会显著影响转染复合物的有效形成;而含有内毒素的质粒对细胞存在很大的毒性作用。抗生素一般对真核细胞无毒,但转染过程中细胞通透性增加,使抗生素进入细胞,从而降低细胞活性,导致转染效率低下。
3. 转染后的筛选时间
转染后筛选不能太早或太晚,因为转染了外源基因的细胞代谢负荷大,增殖较慢,太晚会被没有外源基因转入的细胞所淹没,最终导致筛选不出阳性克隆。一般要在转染 48 h 之后开始加抗生素筛选。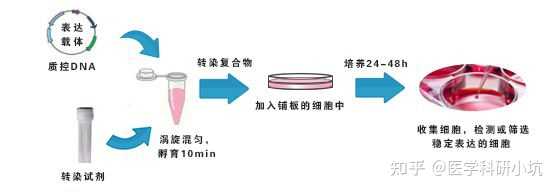
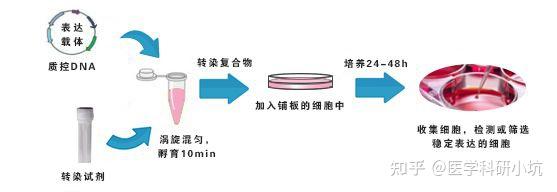
4. DNA 与转染试剂的比例
许多转染方法需要优化 DNA 与转染试剂的比例。不同细胞系转染效率通常不同,细胞系的选择通常根据实验需要确定。在转染前应根据实验要求和细胞特性选择合适的转染试剂,不同转染试剂转染效率差别很大,好的转染试剂能让您事半功倍。



转染(transfection)是细胞在一定条件下主动或被动导入外源DNA片段而获得新的表型的过程。质粒转染对于哺乳细胞蛋白的表达至关重要,转染的方法和步骤直接影响着转染的成功与否。现在一起学习一下质粒转染的具体步骤:以24孔板培养的哺乳动物细胞为例:
1、种细胞;a、贴壁细胞:转染前一天每孔0.5-2×10E5个细胞接种于500ul培养基中,在转染时细胞可长至90-95%汇合度。b、悬浮细胞:在配置转染液前每孔4-8×10E5个细胞接种于500ul培养基中。
2、转染液配置,每孔细胞用量如下:
A. 用50ul无血清培养基稀释质粒DNA,轻轻混匀。B. 取适量合适的质粒转染试剂在50ul无血清培养基中稀释,室温孵育5min。C. 将前两步所稀释的DNA和质粒转染试剂混合,轻轻混匀,室温放置30min。
3、在每孔细胞中加入混合后的转染液,轻轻摇匀。
4、贴壁细胞可在转染4-6h后更换完全培养基,悬浮细胞可在转染后18-48h间进行细胞换液,转染18-48h后可检测基因mRNA水平表达。如果检测蛋白表达,需在转染后48-72h收集细胞样品。


 0
0

材料试剂:胰化蛋白胨,酵母提取物,琼脂,NaCl,NaOH(调pH),氨苄青霉素,卡那霉素,质粒提取试剂盒
仪器:高压灭菌锅,42℃恒温水浴锅,,恒温摇床(37℃,225rpm),无菌培养板,消毒1.5ml离心管,消毒枪头,无菌操作台
实验步骤:
1. LB培养基的配制:
在950 ml去离子水中加入: 胰化蛋白胨 10g 酵母提取物 5g NaCl 10g 摇动容器直至溶质溶解.用5mol/LNaOH调pH至7.0.用去离子水定容至1L.在15psi高压下蒸汽灭菌20min.
固态培养基
LB固体培养基及倒板 :
(1).配制:100mlLB培养基加入1.5g琼脂粉
(2).抗生素的加入:高压灭菌后,将融化的LB固体培养基置与55℃的水浴中,待培养基温度降到55℃时(手可触摸)加入氨苄抗生素(终浓度为50μg/ml),以免温度过高导致抗生素失效,并充分摇匀。
(3).倒板:一般10ml倒1个板子。培养基倒入培养皿后,打开盖子,在紫外下照10-15分钟。
(4).保存:用封口胶封边,并倒置放于4℃保存,一个月内使用。
2. 从-70℃冰箱中取200μl感受态细胞悬液,室温下使其解冻,解冻后立即置冰上。
3. 加入质粒DNA溶液(含量不超过50ng,体积不超过10μl),轻轻摇匀,冰上放置30分钟后。
4. 42℃水浴中热击45s/90s,热击后迅速置于冰上冷却3-5分钟。
5. 向管中加入1ml LB液体培养基(不含Amp),混匀后37℃振荡培养1小时,使细菌恢复正常生长状态,并表达质粒编码的抗生素抗性基因(Ampr )。
6. 将上述菌液摇匀后取100μl 涂布于含Amp的筛选平板上,正面向上放置半小时,待菌液完全被培养基吸收后倒置培养皿,37℃培养16-24小时。
同时做两个对照:
对照组1: 以同体积的无菌双蒸水代替DNA溶液,其它操作与上面相同。此组正常情况下在含抗生素的LB平板上应没有菌落出现。
对照组2: 以同体积的无菌双蒸水代替DNA溶液,但涂板时只取5μl 菌液涂布于不含抗生素的LB平板上,此组正常情况下应产生大量菌落。
大肠杆菌的扩增
1. 从LB平板上挑取新活化的单个菌落,接种于3-5ml LB液体培养基中,37℃下振荡培养12小时左右,直至对数生长后期。将该菌悬液以1:100-1:50的比例接种于100ml LB液体培养基中,37℃振荡培养2-3小时至OD600 =0.5左右。
2. 取上述培养液置于冰中10min,之后转移到已灭菌的50ml离心管中再于冰上放置5min
3. 于4℃离心,2700rmp,10min,弃上清,收集细菌
4. 质粒的提取
准备大肠杆菌质粒提取盒和扩增的带质粒大肠杆菌培养液
5. 质粒鉴定(琼脂糖凝胶电泳)
质粒转染细胞株
材料
细胞株、质粒、培养基、链霉素/青霉素(双抗)、FCS(小牛血清)、PBS(磷酸盐缓冲溶液)、胰酶/EDTA消化液、转染试剂(LipofectamineTM2000)
实验步骤
Ⅰ.准备细胞:
贴壁细胞:转染前 24 h,在 500 µL无双抗完全培养基中接种0.5-2×105个细胞,转染时细胞融合度为80 - 90% 。 ( 注:铺板时要将细胞消化完全混匀,避免细胞堆积生长。)
II .对于每个转染样品,按下面的方法准备:
(1)用50 µL Opti-MEM稀释0.8 µg质粒DNA,轻轻吹吸3 - 5 次混匀,室温下静置5 min。
(2)轻轻颠倒混匀转染试剂,用50 µL Opti-MEM 稀释2.0 µL LipofectamineTM2000,轻轻吹吸3 - 5 次混匀,室温下静置5 min。
(3)混合转染试剂和质粒DNA稀释液,轻轻吹吸 3 - 5 次混匀,室温下静置 20 min。注: 转染复合物一旦形成,应立即加入培养皿中进行细胞转染。
(4)转染复合物加入到24孔细胞板中,100 µL/孔,前后轻摇细胞板混合均匀。
(5)将细胞板置于37℃、5% CO2 培养箱中培养6 h左右,进行换液,换成含10%血清的普通培养基,在37℃,5%CO2孵育箱中继续培养24h左右。
稳定转染细胞株的筛选(各种细胞用什么抗生素筛选呢)
从37℃,5%CO2孵育箱中取出培养皿,弃去含转染试剂的培养基,用PBS冲洗细胞2遍,胰蛋白酶消化,接种1/2的细胞到100 mm培养皿中,加入含500 µg/ml G418的新鲜培养基,每2-3天更换一次新的筛选培养基,每天观察细胞的死亡情况。
当正常细胞完全死亡后,换用新的不含G418的培养基培养。每天观察细胞生长状态。在细胞达到60%汇合率时再用含500 µg/ml G418的培养基筛选一次。
当细胞达到90%以上汇合率时将细胞转移至培养瓶中继续培养(转染后10-12天左右)。以后每隔4-5天再用含500 µg/ml G418的培养基筛选。直到稳定表达转染质粒的细胞达到一定数量后可以收集样品(约转染后15天左右)。以后继续培养时加入的G418浓度降一半。

 0
0


