
2021年7月16日午前3時頃(日本時間)に公開されたAlphaFold ver.2(以下、断りがない限り単純にAlphaFoldと書く)の論文について。
論文は https://www.nature.com/articles/s41586-021-03819-2
実装は https://github.com/deepmind/alphafold
AlphaFold2を使ってみるだけなら(7月19日追記)
Google Colab上で動かせるAlphaFold2を制作してくれた方々がいらっしゃいます。HH-suiteを開発しているSoeding Lab.の方と、RoseTTAFoldの開発の1人であるSergey氏たちの協力によるものですね。

配列をコピペするだけで計算してくれます。
使い方は
1. 上部メニューのランタイムから「ランタイムのタイプを変更」を選び、GPUにセットします(計算速度アップ)
2. 構造予測したいアミノ酸配列を入力欄に入れます。
3. ランタイムメニューから「すべてのセルを実行」を押します。
4. 全行程は8ステップです。現在稼働しているステップは、円マークとその横にあるストップサインで示されます。
このGoogle Colab版は簡易式になっており、以下の点で本家AlphaFold2とは仕様が異なります。使用上の留意点は以下の通り
- MSA取得は本家AlphaFold2がHHblits/hmmerを使っているのに対し、MMseq2を使っています。HHblits/hmmerがBFD, Mgnifyというメタゲノムデータベースに対して行うほど多くの近縁配列を取得してくれるわけではないため、予測精度は低下します。(これは予測したいアミノ酸配列にpsiblastなどでの類縁配列が少ないときに精度の低下を招きますが、類縁配列が多い場合は精度にあまり影響しません)
- 追記: 手持ちのMSAファイルを入力とすることができるように整備中とのこと。
-
本家AlphaFold2は、入力された配列に対して自動的にPDBから最も似ているタンパク質構造をテンプレートとして取得し(これをやるためにpdb70データベースとpdb_mmcifデータベースを本家では利用しています)、そこから構造推論を始めますが、Google Colab版ではテンプレート情報を使っていません。ただこの点については現在追加実装中とのこと。実装されました。 - Google Colab版が用いているSoeding Lab.が作っているMMseqs2は高速で、おそらく1日に20000件以上のリクエストを処理できますが、その計算資源は無限ではありません。利用者が多いと予測に時間がかかるかもです。
-
本家では予測構造を5つデフォルトで計算し、それぞれについてAMBER relaxationの処理を最後に行いますが、Google Colab版では1つのみで行います。複数のモデルが出力できるようになりました。でも効果は小さいかも。
AlphaFold2を使ってみた人々の反応(7月19日追記)
- 多くのタンパク質については結晶構造解析の分子置換に使えるレベルで超高精度の予測結果を返してくれた。
- リンカー領域を入れて2つの配列をつなぐと、複合体もモデリングできる。(←7月19日時点)さらに、小野田さんが発見したリンカーの文字をUやXなど20種類アミノ酸以外の文字にし、32文字以上のリンカーを入れると(suggested by Sergey氏)両ドメインの相対位置を考慮しなくなるのでさらに有効とのこと。特に、ホモ多量体構造予測で効果を発揮する。(7月28日追加)
ただ、この使い方は本来のAlphaFold2の仕様ではない(と思います)し正解できるかどうかはものによるようですが、Sergey氏とBaek氏が試してみたところ、ホモ多量体構造予測については結構な確率で成功するみたいです。この場合、Colab版よりも本家AlphaFold2をローカルにインストールしたバージョンの方が成功率が高いようです。
複合体形成を行うタンパク質A,B,C...が配列データベース上にそれぞれたくさんの類縁配列を持つものならば、その数が多ければ多いほど精度が上がります。このため、データベースを豊富に持ち時間をかけて本気で類縁配列を検索する本家版の方が予測成功率が高いようです。
これはタンパク質の共進化原理に基づいた複合体対象予測として1990年代には知られていた手法のようですが、AlphaFold2の主眼である単量体(モノマー)の構造予測でも、大量の類縁配列から抽出した共進化情報を用いてどのアミノ酸ペアが構造上近位になるかを予測しています。さらなる原理についてはGoogleで「共進化 コンタクト予測」と調べてみてください。
以上のことから、類縁配列が少ない抗原・抗体間のヘテロ複合体予測は難しいんじゃないかなと思います。
- 膜タンパク質はうまく行かない(うまくいくこともある)
一応それらしいきれいな構造は返してくれますが、PDBに近しい構造がない場合、重要な領域は外すことが多いみたいです。
- Intrinsic Disordered Protein (IDP)については効果なし
天然変性タンパク質については予想できません。
インストール手順
マシン環境
十分なマシンスペックが要求されます。具体的には
- 2.5TB以上のSSD/HDD容量 (必須)
- CUDA11に対応しているNVIDIA製GPU(推奨)
- 大容量(32GB以上)のRAM(推奨)
Dockerがあるので究極的にはOSは何でも良いはずですが、macOSでは後述の理由で現実的には厳しいでしょう。NVIDIA製GPUが利用できる都合上LinuxやWindowsの方が便利です。最後のAlphaFold2本体による構造推論部分はGPUがあるとn倍速になりますが、そこの部分の計算に限って言えば、気長に待てばmacOSにおいても現実的な範囲内で計算できそうです(未検証ですが)。ただ、結局2.5TB以上のSSD/HDD容量は必須なのでどのみちmacOSには厳しいです。外付けHDD上で動かすという手も考えられますが、構造推論の全行程の最初にあるHH-suite/hhblits処理が非効率になり、計算速度が数倍落ちます。
AlphaFoldははじめHH-Suiteを使ってホモログ検索をしますが、このとき大容量のRAM(いわゆるメモリのこと)があればあるほど計算が速くなり、次点でRead/Write速度が速いSSDが望ましいです。おすすめはディスク領域が2.5TB以上あるM.2のSSDまたは内蔵HDD入りのメモリ32GB(64GBあればなお良い)+RTXの2060以上か3060以上のGPUが備わったゲーミングデスクトップパソコン(新品ならだいたい30万円前後くらい)での動作です。
AlphaFold2の構造推論処理はHH-suiteによるMSA(Multiple sequence alignment)取得が計算時間の8〜9割、Deep Learningによる学習済みモデルを用いた構造推論が1割を占めるので、速さを求めるなら大容量SSD, 大容量RAMがあるマシンを使いましょう。HH-suiteの計算時間向上についてはHH-suiteのWikiが詳しいです。構造予想部分に使うことになるGPUもRTX2080や3090といったハイエンドのものでなくても現実的な時間内で動きそうです。
Dockerのインストール
aptを使ったインストール
Ubuntu20.04の場合、以下の手順で導入します。
# aptのアップデート
sudo apt -y update
# aptがHTTPS経由でパッケージを使用できるようにするいくつかの必要条件パッケージをインストール
sudo apt -y install apt-transport-https ca-certificates curl software-properties-common
# 公式DockerリポジトリのGPGキーをシステムに追加
curl -fsSL https://download.docker.com/linux/ubuntu/gpg | sudo apt-key add -
sudo add-apt-repository "deb [arch=amd64] https://download.docker.com/linux/ubuntu $(lsb_release -cs) stable"
# 追加されたリポジトリからDockerパッケージでパッケージデータベースを更新
sudo apt -y update
# docker-ceのインストール
sudo apt-get install -y docker-ce
以上でDockerがインストールされます。dockerを確認します。
$ docker version
Client: Docker Engine - Community
Version: 20.10.7
API version: 1.41
Go version: go1.13.15
Git commit: f0df350
Built: Wed Jun 2 11:56:38 2021
OS/Arch: linux/amd64
Context: default
Experimental: true
Got permission denied while trying to connect to the Docker daemon socket at unix:///var/run/docker.sock: Get http://%2Fvar%2Frun%2Fdocker.sock/v1.24/version: dial unix /var/run/docker.sock: connect: permission denied
Dockerがインストールされ、デーモンが起動し、プロセスがプート時に起動できるようになりました。実行されていることを確認します。
$ systemctl status docker
● docker.service - Docker Application Container Engine
Loaded: loaded (/lib/systemd/system/docker.service; enabled; vendor preset: enabled)
Active: active (running) since Fri 2021-07-16 15:52:09 JST; 9min ago
TriggeredBy: ● docker.socket
Docs: https://docs.docker.com
Main PID: 298145 (dockerd)
Tasks: 32
Memory: 50.7M
CGroup: /system.slice/docker.service
└─298145 /usr/bin/dockerd -H fd:// --containerd=/run/containerd/containerd.sock
Active: active (running)になっていれば成功。正しく起動できていない場合はここが赤色で表示されます。
Dockerを非rootユーザーでも使えるようにする
rootでなく一般ユーザでdockerを実行する場合には、Linuxの場合dockerグループに所属している必要があります。このため任意のユーザーを管理者権限でひとりひとりdocker groupに入れてあげます。例えばmoriwakiユーザーをdockerグループに入れたい場合は
sudo usermod -aG docker moriwaki
と実行する必要があります。
ここまで終わったら一度ログアウトし、再びログインした後にコマンドdocker run hello-worldを実行します。
$ docker run hello-world
Unable to find image 'hello-world:latest' locally
latest: Pulling from library/hello-world
b8dfde127a29: Pull complete
Digest: sha256:df5f5184104426b65967e016ff2ac0bfcd44ad7899ca3bbcf8e44e4461491a9e
Status: Downloaded newer image for hello-world:latest
Hello from Docker!
This message shows that your installation appears to be working correctly.
このようなメッセージが表示されていれば準備OKです。
NVIDIA Container Toolkitのインストール
Dockerをインストールし終わったあとで、公式の記述 https://docs.nvidia.com/datacenter/cloud-native/container-toolkit/install-guide.html を元にインストールします。
distribution=$(. /etc/os-release;echo $ID$VERSION_ID) \
&& curl -s -L https://nvidia.github.io/nvidia-docker/gpgkey | sudo apt-key add - \
&& curl -s -L https://nvidia.github.io/nvidia-docker/$distribution/nvidia-docker.list | sudo tee /etc/apt/sources.list.d/nvidia-docker.list
この後、nvidia-docker2 packageをインストールします。
sudo apt-get update
sudo apt-get install -y nvidia-docker2
インストールを完了するために、dockerのデーモンを再起動します。
sudo systemctl restart docker
ここで正しく動作しているかどうかを以下のコマンドで確認します。
sudo docker run --rm --gpus all nvidia/cuda:11.0-base nvidia-smi
nvidia-smiコマンドを実行したときのように、GPU情報が表示されれば成功。こんな感じの画面。
$ docker run --rm --gpus all nvidia/cuda:11.0-base nvidia-smi
Fri Jul 16 08:24:07 2021
+-----------------------------------------------------------------------------+
| NVIDIA-SMI 460.84 Driver Version: 460.84 CUDA Version: 11.2 |
|-------------------------------+----------------------+----------------------+
| GPU Name Persistence-M| Bus-Id Disp.A | Volatile Uncorr. ECC |
| Fan Temp Perf Pwr:Usage/Cap| Memory-Usage | GPU-Util Compute M. |
| | | MIG M. |
|===============================+======================+======================|
| 0 GeForce RTX 3090 Off | 00000000:0D:00.0 Off | N/A |
| 54% 35C P0 26W / 350W | 0MiB / 24265MiB | 0% Default |
| | | N/A |
+-------------------------------+----------------------+----------------------+
+-----------------------------------------------------------------------------+
| Processes: |
| GPU GI CI PID Type Process name GPU Memory |
| ID ID Usage |
|=============================================================================|
| No running processes found |
+-----------------------------------------------------------------------------+
エラーだとこんな感じで表示されます。
docker: Error response from daemon: could not select device driver "" with capabilities: [[gpu]].
Alphafoldのリポジトリのクローン
GitHubからAlphaFoldをダウンロードしてきます。
git clone https://github.com/deepmind/alphafold.git
cd alphafold
run_dockerファイルの編集
リポジトリをクローンしたらさっそくいくつかのパラメータを変えておきます。docker/run_docker.pyファイルを開いて以下の部分を編集します。
#### USER CONFIGURATION ####
# Set to target of scripts/download_all_databases.sh
DOWNLOAD_DIR = 'SET ME'
# Name of the AlphaFold Docker image.
docker_image_name = 'alphafold'
# Path to a directory that will store the results.
output_dir = '/tmp/alphafold'
これはユーザーそれぞれが好きなように設定しておきます。例えば私はこうしました。
#### USER CONFIGURATION ####
# Set to target of scripts/download_all_databases.sh
DOWNLOAD_DIR = '/data/moriwaki/database'
# Name of the AlphaFold Docker image.
docker_image_name = 'alphafold'
# Path to a directory that will store the results.
output_dir = '/data/moriwaki/alphafold_out'
ここで指定したDOWNLOAD_DIR変数のディレクトリは、後のデータベースの準備のときのディレクトリ名と同一にします。
また、ここで設定したoutput_dirは存在していないとAlphaFold2の動作時にエラーが起きるので、ディレクトリを作成しておきます(重要)。
$ mkdir -p /data/moriwaki/alphafold_out
上のスクリプトで設定したアウトプット用ディレクトリが作成されました。
Dockerfileの修正
たぶんいずれ公式の方で修正されるとは思いますが、Alphafold初公開直後の時点ではこのdocker/Dockerfileを修正してあげないとうまく動かないです。
7月23日、修正されたと思います。以下の操作は必要ありません
https://github.com/deepmind/alphafold/issues/11 を参考にして修正してください。
データベースの準備
AlphaFoldを実行するためには以下の遺伝的(配列)データベースが必要なのでダウンロードしておきます。
公式GitHubがscripts/download_all_data.sh に全部一括でダウンロードしてくれるスクリプトを置いてくれているので、それを使えば全部いい感じにやってくれます。これには8〜12時間かかるとされていますが日本からだともっとかかります。実際にはこのスクリプトを実行する前にいくつか変更を加えておいた方がこの辺の準備を早く行えます(後述)。
このスクリプトを使う場合は先に強力なダウンロードコマンドのrsyncとaria2(aria2c)をインストールしておく必要があります。インストールはUbuntu20.04の場合
sudo apt -y install rsync aria2
でできます。他のOSの場合は割愛。
次に先述したデータベースのインストールディレクトリ/data/moriwaki/databaseを用いて、そのディレクトリにデータベースをダウンロードしていきます(が、下記の内容を含めてこの項全体をよく読んでから実行することをおすすめします)
INSTALLDIR="/data/moriwaki/database"
scripts/download_all_data.sh ${INSTALLDIR}
とすることで、その指定したディレクトリにデータベースを順々にダウンロードしてくれます。
このスクリプトの実態は同じディレクトリにある他7つのダウンロード実行スクリプト
download_alphafold_params.shdownload_bfd.shdownload_mgnify.shdownload_uniref90.shdownload_uniclust30.shdownload_pdb70.shdownload_pdb_mmcif.sh
これらを順番に実行するものとなっており、これらのスクリプトを並列で実行した方が早いです。並列で実行したい場合は
INSTALLDIR="/data/moriwaki/database"
nohup scripts/download_alphafold_params.sh ${INSTALLDIR} > dl_params.out &
nohup scripts/download_bfd.sh ${INSTALLDIR} > dl_bfd.out &
...(以下略)
みたいな感じでバックグラウンド処理させると良い。dl_xxx.outファイルにそのダウンロードの記録が残るような仕組みになっています。また、各コマンドの先頭にnohupをつけて開始させると、ssh接続を閉じたりターミナルを閉じたりしても処理が続行されるので便利です。もしその処理を止めたい場合はps auxでプロセス番号(PID)を調べてからkill -9 <PID>とします。
以下は上記コマンドを実行する前に知っておくと便利なTIPS。
download_alphafold_params.sh
学習済みのalphafoldのパラメータたちをダウンロードします。注意点として、AlphaFoldのコードはすべてApache 2.0 Licenseであるため完全に自由に改変していいし商用利用も可能ですが、これでダウンロードされるパラメータファイルはCC BY-NC 4.0 Licenseなので、使用時には適切なクレジット表示をしなければならず、営利目的での利用は禁止されています。
download_bfd.sh
AlphaFoldの構造予測プロセスの初期段階で用いるHH-suite ver.3.3で、クエリ配列に対するリモートホモログ配列を取得するときに使う追加データベースのBFDをダウンロードする。https://bfd.mmseqs.com/ からダウンロード可能。下記のuniclust30データベースに加えてUniprot/Tremble, Swiss-protといった完全な配列だけでなく、メタゲノム配列を加えてさらに配列空間を増強することで共進化情報の精度を大幅に上げることに成功したという2017年の論文以降、その要因でHH-suiteチームも公式にメタゲノム配列を加えたデータベースBFDを提供し定期的に更新して配信してくれています。
しかし、非常に重い。2.0TBくらいある。 これが理由で大容量HDDが要求されます。このデータベースは上記の通り使わなくても原理上は動かすことは可能だが、あった方が大変よろしい。
解凍後の当該ディレクトリはこのようなファイル構成になります。
[/data/moriwaki/database/bfd] $ ls -lt
-rwxr-xr-x 1 moriwaki moriwaki 1688518403 3月 5 2019 bfd_metaclust_clu_complete_id30_c90_final_seq.sorted_opt_cs219.ffindex
-rwxr-xr-x 1 moriwaki moriwaki 16814534465 3月 5 2019 bfd_metaclust_clu_complete_id30_c90_final_seq.sorted_opt_cs219.ffdata
-rw------- 1 moriwaki moriwaki 1555724298013 3月 4 2019 bfd_metaclust_clu_complete_id30_c90_final_seq.sorted_opt_a3m.ffdata
-rw------- 1 moriwaki moriwaki 326882934659 3月 3 2019 bfd_metaclust_clu_complete_id30_c90_final_seq.sorted_opt_hhm.ffdata
-rw------- 1 moriwaki moriwaki 129631317 3月 3 2019 bfd_metaclust_clu_complete_id30_c90_final_seq.sorted_opt_hhm.ffindex
-rw------- 1 moriwaki moriwaki 1817298807 3月 3 2019 bfd_metaclust_clu_complete_id30_c90_final_seq.sorted_opt_a3m.ffindex
download_uniclust30.sh
AlphaFoldの構造予測プロセスの初期段階で用いるHH-suite ver.3.3で、クエリ配列に対するリモートホモログ配列を取得するときに必須のデータベースです。http://wwwuser.gwdg.de/~compbiol/uniclust/2018_08/ に置かれているuniclust30_2018_08_hhsuite.tar.gzファイルをダウンロードして展開する処理を行います。しかし、このデータベースは2018年版なので、http://wwwuser.gwdg.de/~compbiol/uniclust/2021_06/ に置かれている最新版の UniRef30_2021_06_hhsuite.tar.gz を用いてもOKです。HH-suite本家ウェブサーバーではこちらがデフォルトになっている。一応、論文に合わせるなら2018_08版を使う方が無難かも?
lrwxrwxrwx 1 moriwaki moriwaki 29 Oct 18 18:01 uniclust30_2018_08_hhm_db -> uniclust30_2018_08_hhm.ffdata
lrwxrwxrwx 1 moriwaki moriwaki 29 Oct 18 18:01 uniclust30_2018_08_a3m_db -> uniclust30_2018_08_a3m.ffdata
-rw-r--r-- 1 moriwaki moriwaki 767 Oct 12 07:13 uniclust30_2018_08_md5sum
-rw-r--r-- 1 moriwaki moriwaki 9427452 Oct 12 07:10 uniclust30_2018_08_hhm_db.index
-rw-r--r-- 1 moriwaki moriwaki 436453157 Oct 12 07:10 uniclust30_2018_08_a3m_db.index
-rw-r--r-- 1 moriwaki moriwaki 4044015407 Oct 12 07:09 uniclust30_2018_08.cs219
-rw-r--r-- 1 moriwaki moriwaki 19 Oct 12 07:09 uniclust30_2018_08.cs219.sizes
-rw-r--r-- 1 moriwaki moriwaki 8167108 Oct 12 07:06 uniclust30_2018_08_hhm.ffindex
-rw-r--r-- 1 moriwaki moriwaki 14171782777 Oct 12 07:06 uniclust30_2018_08_hhm.ffdata
-rw-r--r-- 1 moriwaki moriwaki 375805833 Oct 12 07:05 uniclust30_2018_08_a3m.ffindex
-rw-r--r-- 1 moriwaki moriwaki 69490273138 Oct 12 07:05 uniclust30_2018_08_a3m.ffdata
-rw-r--r-- 1 moriwaki moriwaki 356545673 Oct 12 07:02 uniclust30_2018_08_cs219.ffindex
-rw-r--r-- 1 moriwaki moriwaki 3860466139 Oct 12 07:02 uniclust30_2018_08_cs219.ffdata
(補足)
もしuniclust30ではなくUniRef30_2021_06_hhsuite.tar.gz を用いたい場合はdownload_uniclust30.shスクリプトの中身を適宜書き換えてください。そしてdocker/run_docker.pyの71行目あたりにある記述を
# Path to the Uniclust30 database for use by HHblits.
uniclust30_database_path = os.path.join(
- DOWNLOAD_DIR, 'uniclust30', 'uniclust30_2018_08', 'uniclust30_2018_08')
+ DOWNLOAD_DIR, 'UniRef30_2021_06', 'UniRef30_2021_06')
というように変更します。これで問題なく動作します。UniRef30_2021_06を利用した場合のファイル構成は以下の通り。
-rw-r--r-- 1 moriwaki moriwaki 379 Jul 19 11:35 UniRef30_2021_06.md5sums
-rw-r--r-- 1 moriwaki moriwaki 40534690979 Jul 19 11:30 UniRef30_2021_06_hhm.ffdata
-rw-r--r-- 1 moriwaki moriwaki 22673255 Jul 19 11:30 UniRef30_2021_06_hhm.ffindex
-rw-r--r-- 1 moriwaki moriwaki 794584753 Jul 19 11:29 UniRef30_2021_06_a3m.ffindex
-rw-r--r-- 1 moriwaki moriwaki 171469032756 Jul 19 11:29 UniRef30_2021_06_a3m.ffdata
-rw-r--r-- 1 moriwaki moriwaki 716343145 Jul 19 11:25 UniRef30_2021_06_cs219.ffindex
-rw-r--r-- 1 moriwaki moriwaki 7228866880 Jul 19 11:24 UniRef30_2021_06_cs219.ffdata
download_mgnify.sh
mgnifyのダウンロード。特に言うことなし。というかmgnifyについてよく調べてないです……。いずれ調べて書きます。
download_uniref90.sh
タンパク質のアミノ酸配列と機能情報を高品質で包括的に提供することを目指すUniprotデータベースに置かれてあるUniProt Reference Clusters (UniRef)の配列。UniRef100に含まれるアミノ酸配列をクラスタリングすることで構築されており、各クラスタは最長の配列(シード配列)に対して90%以上の相同性を持ち、かつ80%以上のオーバーラップを持つ配列で構成されています。
UniRef90 is built by clustering UniRef100 sequences such that each cluster is composed of sequences that have at least 90% sequence identity to, and 80% overlap with, the longest sequence (a.k.a. seed sequence).
解凍されたuniref90.fastaファイルの中には大量のアミノ酸配列がFASTAフォーマットでテキスト形式で書かれており、これとUniclust30 + BFDデータベースとHH-suiteソフトウェアを用いることで、uniref90.fastaファイルの中からクエリ配列に対するリモートホモログを検出します。
download_pdb70.sh
HH-Suiteが提供するデータベースの1つで、Protein Data Bankに登録されている構造の配列をクラスタリングしたもの。毎週1回アップデートされています。http://wwwuser.gwdg.de/~compbiol/data/hhsuite/databases/hhsuite_dbs/ から最新版をダウンロード可能。
元はHH-Suiteのチームが2005年に作成した構造予測プログラムHHPredのために作られているデータベース。テンプレートベースの構造予測としては非常に高速で強力です。AlphaFoldはこれと似たアルゴリズムでテンプレートとして計算されたタンパク質立体構造を構造予測の出発点としているみたいです。
解凍後はこんな感じ。
[/data/moriwaki/database/pdb70] $ ls -lt
-rw-r--r-- 1 moriwaki moriwaki 410 Jul 8 23:52 md5sum
-rw-r--r-- 1 moriwaki moriwaki 3875841099 Jul 8 23:47 pdb70_hhm.ffdata
-rw-r--r-- 1 moriwaki moriwaki 2066799 Jul 8 23:47 pdb70_hhm.ffindex
-rw-r--r-- 1 moriwaki moriwaki 63005744825 Jul 8 23:46 pdb70_a3m.ffdata
-rw-r--r-- 1 moriwaki moriwaki 2245399 Jul 8 23:46 pdb70_a3m.ffindex
-rw-r--r-- 1 moriwaki moriwaki 24942439 Jul 8 23:08 pdb70_cs219.ffdata
-rw-r--r-- 1 moriwaki moriwaki 1715919 Jul 8 23:08 pdb70_cs219.ffindex
-rw-r--r-- 1 moriwaki moriwaki 8204591 Jul 7 10:01 pdb70_clu.tsv
-rw-r--r-- 1 moriwaki moriwaki 23827536 Jul 7 10:00 pdb_filter.dat
AlphaFoldのデータベースのタイムスタンプは2020-05-13となっていますが、特にこだわりがない場合は最新のものを使う方が良いでしょう。
download_pdb_mmcif.sh
Protein Data Bank(PDB)のmmCIFフォーマットの構造データをまるごとローカルにミラーリングするスクリプト。
しかし日本から利用する場合はrsyncする対象のウェブサイトを日本のミラーサーバーに変更してから実行した方が圧倒的に速い。そこで44〜45行目の部分を
- rsync --recursive --links --perms --times --compress --info=progress2 --delete --port=33444 \
- rsync.rcsb.org::ftp_data/structures/divided/mmCIF/ \
+ rsync --recursive --links --perms --times --compress --info=progress2 --delete \
+ ftp.pdbj.org::ftp_data/structures/divided/mmCIF/ \
このように書き換えてから実行することを強くおすすめします。
実行すると指定したデータベースのダウンロードディレクトリ以下にpdb_mmcif/raw/xxというディレクトリが生成され(xxは4文字のPDB IDの2〜3文字め)、そこにまず圧縮されたファイルxxxx.cif.gzがダウンロードされます。続いて全ファイルのgzip圧縮を解凍する処理が始まります。
AlphaFold実行の手順
最も簡単な方法はdocker/run_docker.pyを使ってDocker環境で実行することです。
-
docker/run_docker.pyファイルの中にあるDOWNLOAD_DIRという文字を、自身の環境で設置したデータベースのディレクトリのPATHに書き換えます。例えば/data/moriwaki/databaseみたいな感じ。
# Set to target of scripts/download_all_databases.sh
- DOWNLOAD_DIR = 'SET ME'
+ DOWNLOAD_DIR = '/data/moriwaki/database'
-
Dockerイメージを構築します。
docker build -f docker/Dockerfile -t alphafold .もしbuildした後にDockerfileの設定をし忘れていていることに気付いて設定を変更する必要があった場合は、
docker imagesでalphafold2のIMAGE IDを取得後、docker rmi <IMAGE ID> -fとしてそのイメージを削除してからbuildしなおしましょう。詳しくはdockerのimageの削除などでググってください。 -
run_docker.pyの実行に必要なPythonパッケージをインストールします。(場合によってはvirtualenvなどで仮想環境を作ってシステムのPython環境とコンフリクトしないようにしておくのも手でしょう。)pip3 install -r docker/requirements.txt -
構造予測したいアミノ酸配列が入ったFASTAフォーマットのファイルを作成し、そのファイルへのPATHを指定して
run_docker.pyスクリプトを実行します。もし予測したい構造がすでにPDBに登録されており、それをテンプレートとして用いることを避けたい場合は、max_template_dateというオプションを指定することで、それ以前のPDBデータ公開日の構造をもとに構造予測することができます。例えばCASP14のターゲットのひとつT1050についてはpython3 docker/run_docker.py --fasta_paths=/path/to/T1050.fasta --max_template_date=2020-05-14またデフォルトではAlphaFoldはすべての取得可能なGPUデバイスを利用して計算しようとします。一部だけ使いたい場合は、コンマで区切られたGPUのUUIDまたはindexのリストを指定して
--gpu_devicesフラグに入力します。詳細は
GPU enumerationを読んでください。 -
AlphaFoldの計算速度と精度のトレードオフを
--preset=full_dbsまたは--preset=casp14オプションを付けることで指定できます。それぞれのプリセットは以下の通り- casp14: CASP14で使われた設定と同じ高精度のもの。すべての配列データベースを用いて8アンサンブルで実行する。
-
full_dbs: こっちがデフォルト設定のはず。こちらは
casp14プリセットよりも8倍速く計算でき、モデリングクオリティの低下もかなり小さい(CASP14でのドメインを対象に、平均してGDTスコアでわずか-0.1)。すべての配列データを用いるがアンサンブルは伴わない。
caps14プリセットで実行する場合は以下のようにする。python3 docker/run_docker.py --fasta_paths=T1050.fasta --max_template_date=2020-05-14 --preset=casp14
いざ実行
構造予測したいアミノ酸配列が含まれたFASTAフォーマットのテキストファイルを用意します。例えばここでは遺伝子工学でよく使われる(使われていた?)Type-2 制限酵素 XhoIの構造予測をしてみたいと考えます。この酵素はまだ構造が明らかになっていません。
Uniprotでは https://www.uniprot.org/uniprot/Q9KVZ7 に情報がまとめられており、FASTAファイルは https://www.uniprot.org/uniprot/Q9KVZ7.fasta からダウンロードできます。
>sp|Q9KVZ7|T2X1_XANVA Type-2 restriction enzyme XhoI OS=Xanthomonas vasicola OX=56459 PE=3 SV=1
MALDLAEYDRLARLGVAQFWDGRSSALENDEERSQGGERSGVLGGRNMDGFLAMIEGIVR
KNGLPDAEVCIKGRPNLTLPGYYRPTKLWDVLVFDGKKLVAAVELKSHVGPSFGNNFNNR
AEEAIGTAHDLATAIREGILGDQLPPFTGWLILVEDCEKSKRAVRDSSPHFPVFPDFKGA
SYLTRYEVLCRKLILKGFTPRPQSLLRPALRVLGATIASFRKPRVCAHLRHGWPAMYPVG
QSRIRVNF
FASTAファイルフォーマットとは、一行目に>で始まるコメント行(何を書いても良い)、二行目以降がすべてアミノ酸配列(改行可能)となっているファイル形式です。このルールさえ守っていれば自前でFASTAファイルを書いてもOKです。これをQ9KVZ7.fastaとしてホームディレクトリ上に保存し(どこでも良い)、これについて構造予測してみます。
Alphafoldの計算時間は最低1〜3時間以上かかると思いますが、nohupコマンドをつけるとリモート先でログアウトした後でも計算し続けてくれるので便利です。alphafoldのディレクトリ上にcdで移動して、例えばホームディレクトリ上に置いてあるQ9KVZ7.fastaについて構造予測したい場合はこんな感じ。
$ nohup python3.8 docker/run_docker.py \
--fasta_paths=${HOME}/Q9KVZ7.fasta \
--max_template_date=2021-07-14 \
--preset=full_dbs > nohup1.out &
計算結果は、Dockerfileのファイルの中の変数output_dirで指定したディレクトリに出力されます。標準出力&エラーメッセージはnohup1.outファイルに書き込まれます(この名前は自由に指定可)。
--max_template_dateの引数は必須で、指定した日までにPDBで公開された構造をAlphaFoldの構造予測のテンプレート(初期構造)として利用してくれます。この機能により、正解構造がPDBにある状態でもそれ以外の構造を元にテンプレートを選択してくれるようです。(それとは別にDeep Learningで用いる学習モデル自体は2020年5月14日までの全PDBデータを何らかの形で利用して学習しているみたいなのですが)。
--preset=で指定するオプションはfull_dbsかcasp14の二択で、通常はfull_dbsを使うと良いでしょう。casp14は構造予測コンテスト用に8倍時間をかけて精度を高めてくれるオプションですが、効果の差は小さいらしいので、まずはfull_dbsを指定して不満だったらcasp14を指定し直して試してみる、といった感じでしょうか。
結果
output_dirで指定したディレクトリに作られたrelaxed_model_*.pdbが予測構造結果です。驚きの予測精度を自身の目で確認してみましょう。
残基数380くらいのタンパク質で計算したところ計算時間129分となり(--preset=full_dbsの場合)、このうちhhblitsによるMSA取得が116分、最後のAlphaFold2本体による構造推論部分が13分でしたので、"GPUなしの計算機"でも気長に待てば現実的な範囲内で終わりそうです。ただいずれにしろそこそこ新しいCPUと最低32GBのRAMはほしいところです。
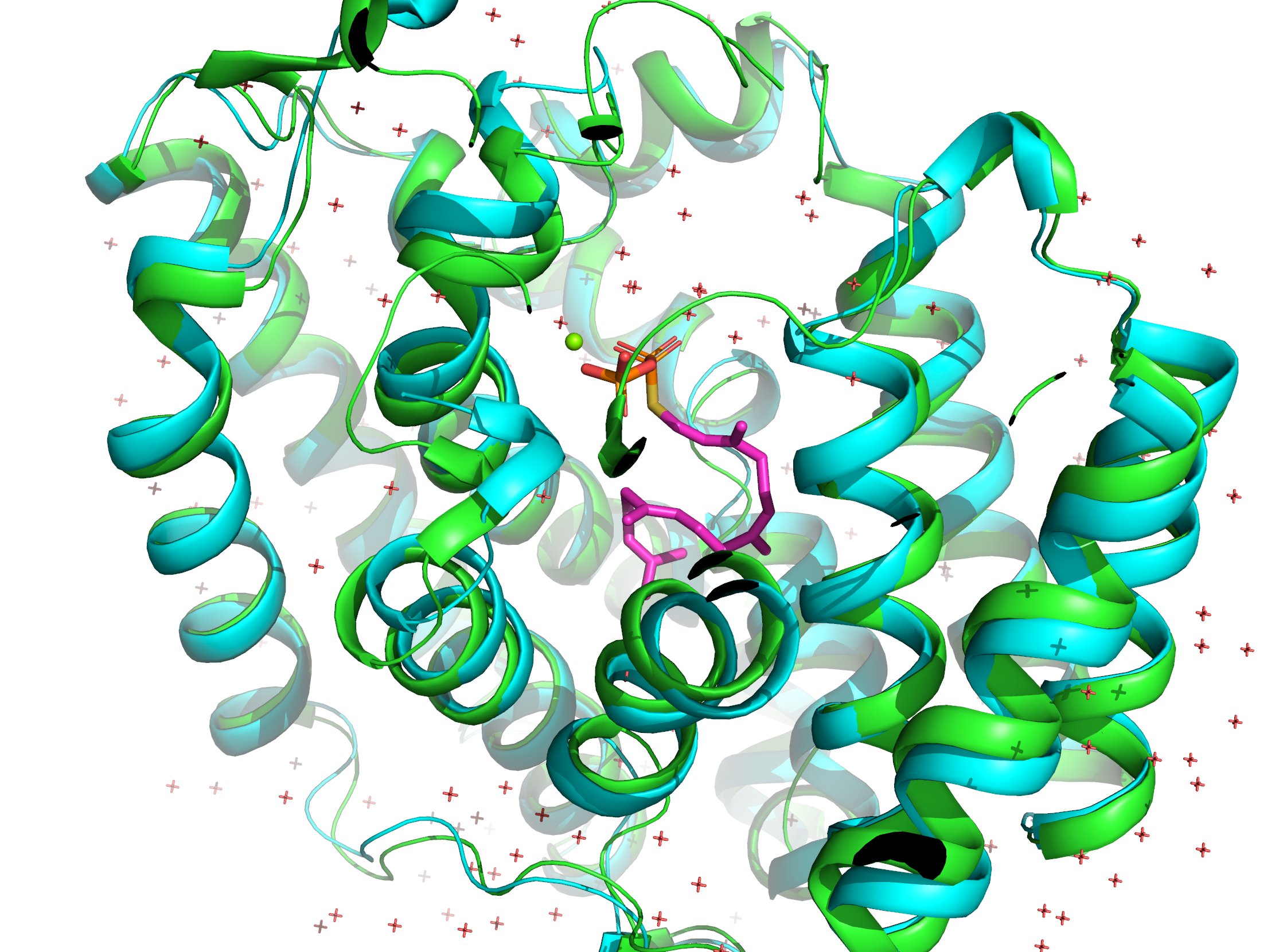
AlphaFoldと同日にScience誌で発表されたRoseTTAFoldとの性能比較を、PDB: 5GUEで行ってみました。AlphaFold (1枚目) とRoseTTAFold (2枚め)です。両者とも正解構造は知らない状態で予測しました(はず)。緑が予想構造で青が正解構造です。
RoseTTAFoldの正解構造からのずれを表すRMSD値は1.474Åでしたが、AlphaFold2は0.362Åでした。圧倒的精度!

参考資料(8月6日追記)
- AlphaFold2解体新書プロジェクト
- 英語の解説動画 Martin Steinegger氏はAlphaFold論文の著者の1人





コメント