

Does Glyphosate Acting as a Glycine Analogue Contribute To ALS?
Wendy A. Morley2, Michael J. Hadden3, Martin C. Michener4

Abstract
Amyotrophic Lateral Sclerosis (ALS) is a fatal neurodegenerative disease involving several protein mutations in glycine-rich regions with limited treatment options. 90 - 95% of all cases are non-familial with epidemiological studies showing a significant increased risk in glyphosate-exposed workers. In this paper, we propose that glyphosate, the active ingredient in Roundup®, plays a role in ALS, mainly through mistakenly substituting for glycine during protein synthesis, disruption of mineral homeostasis as well as setting up a state of dysbiosis. Mouse models of ALS reveal a pre-symptomatic profile of gut dysbiosis. This dysbiotic state initiate a cascade of events initially impairing metabolism in the gut, and, ultimately, through a series of intermediate stages, leading to motor neuron axonal damage seen in ALS. Lipopolysaccharide, a toxic by-product of dysbiosis which contributes to the pathology, is shown to be statistically higher in ALS patients. In this paper we paint a compelling view of how glyphosate exerts its deleterious effects, including mitochondrial stress and oxidative damage through glycine substitution. Furthermore, its mineral chelation properties disrupt manganese, copper and zinc balance, and it induces glutamate toxicity in the synapse, which results in a die-back phenomenon in axons of motor neurons supplying the damaged skeletal muscles.
Introduction
Amyotrophic Lateral Sclerosis (ALS) is a fatal neurodegenerative disease characterized by a progressive loss of motor neurons along with muscle atrophy. ALS is an adult onset disease, usually manifested first by weakness in the arms or legs and proceeding on to paralysis and death due to respiratory failure, often within five years of diagnosis[1,2]. The lifetime risk is between 1 in 400 and 1 in 1000, making it the most common motor neuron disease[3-5]. As of now there is no known treatment or cure. Recent evidence points to an important role in gut dysbiosis in early stages of the disease. A seminal paper by Wu et al., published in 2015, examined alterations in gut microbes and associated metabolites in a mouse model for ALS, specifically targeting disruptions that preceded the onset of disease manifestations[6]. This ALS mouse model involves the expression of a mutant form of human superoxide dismutase 1 (SOD1), where glycine at residue 93 is replaced with alanine. The study revealed that young mice expressing human G93A SOD1 exhibited damaged tight junction structure and increased gut permeability. This was associated with reduced expression of tight junction proteins Zonulin occludens-1 and E-cadherin, leading to impaired gut barrier function. Paneth cells were also abnormal, with decreased expression of the antimicrobial peptide defensin 5 alpha. Reduced levels of butyrate-producing intestinal microbes such as Butyrivibrio fibrisolvens and Fermicus and increased expression of the inflammatory cytokine IL-17, as well as reduced levels of autophagic lysozyme 1 (leading to a reduced ability to clear misfolded proteins) were further markers of a disrupted microbiome. All of these features appeared when the mice were only 2 months old, before any symptoms of ALS had yet developed.
Only about 5 - 10 % of the cases of ALS can be linked to known genetic defects (familial cases); the rest are idiopathic or sporadic (sALS). About 20 % of the familial cases are linked to mutations in SOD1[7]. There is a striking pathological and clinical similarity between familial and idiopathic disease, leading researchers to believe that the mouse SOD1 model may be representative of the sporadic cases as well as the familial ones. SOD1’s main function is to convert superoxide, produced as a toxic by-product of mitochondrial oxidative phosphorylation, to water or hydrogen peroxide. The deleterious effect of mutant SOD1 however is not due to a loss of this function; most mutant forms exhibit full activity or even enhanced activity levels[3]. While there are more than 180 identified mutations of SOD1 in humans, three have been extensively characterized in transgenic mouse models, all of which involve substitutions for a conserved glycine residue in the original protein (SOD1G85R, SOD1G37R, and SOD1G93A). Often the mutant protein is expressed in the mouse genome at levels that are several-fold higher than the levels of endogenous SOD1[8]. Substitution of arginine for a highly conserved glycine (G10R) in a patient with fALS strongly destabilized the protein secondary structure, leading to intracellular aggregates[9].
Impaired mitochondrial function has been linked to ALS, particularly related to Cytochrome c Oxidase (CcO)[10,11], and mutant SOD1 has been shown to suppress CcO activity[12]. Intriguingly, both CcO and SOD1 depend on copper as a catalyst. Studies on G93A-SOD1 mice showed that the mutant mice exhibited a decrease in mitochondrial respiration specific to complex IV, and this defect was tied to impaired cytochrome c oxidation, and occurred long before any overt symptoms appeared[13]. The amount of CcO on the inner mitochondrial membrane was reduced, and this was correlated with increased peroxidation of lipids, including cardiolipin. One explanation is that over expression of mutant SOD1 led to a decrease in the bioavailability of copper to CcO, leading to both impaired complex IV oxidative phosphorylation and the excessive expression of reactive oxygen species. Another proposed theory is that mutant SOD1 induces conformational changes that facilitate the interactions of catalytic copper with peroxynitrite or hydrogen peroxide to generate toxic free radicals which then damage cellular proteins and lipids[12].
Remarkably, a variant of SOD1 that lacks all crucial residues needed for coordinate binding of Cu induces an ALS-like disease in mice that is indistinguishable from the disease phenotypes of mice expressing other human SOD1 mutant forms[14]. The disease manifestation appears as the accumulation of misfolded high-molecular-weight SOD1 aggregates. This feature is also observed in the glycine-substituted mouse models. This suggests that a copper deficiency or impairment in copper binding may be a factor in the disease process, leading to oxidative and nitrosative stress due to free copper along with misfolded copper-deprived versions of both SOD1 and CcO.
A study by Beckman et al. examined mice that over expressed both human SOD and human copper chaperone for superoxide dismutase (CCS)[15]. These mice die of ALS at an accelerated pace, probably because CCS sequestered the mouse’s supply of copper, inducing copper deficiency in the spinal cord. The depletion of copper affected both SOD1 and CcO function. A study from 2016 shows almost conclusively that impaired copper supply to SOD1 and to CcO is a strong factor in the disease[16]. These authors showed that a copper chelator, CuATSM, dissolved in dimethyl sulfoxide and dribbled onto the pup’s neck, was absorbed into the skin and became a bioavailable supply of copper to the central nervous system. This treatment had an effect within a few hours to significantly increase mobility in diseased mice, who suffered from both a SOD1 mutation and excessive expression of CCS. The general model that is emerging is that mutant SOD1 induces both protein misfolding and oxidative stress perhaps mediated by free copper that leads to mitochondrial dysfunction, along with impaired transport along axons leading to neuroinflammation and apoptosis of motor neurons[17].
The accumulation of misfolded proteins in ALS is not limited to SOD1. Two nuclear RNA/DNA binding proteins, TAR (transactive response) DNA-binding protein 43 (TDP-43) and FUsed in Sarcoma (FUS), are both found to be genetically linked to ALS[18]. TDP-43 is nearly always found in aggregates with ubiquitin in ALS inclusions, both familial and sporadic[3,19]. In fact, remarkably, it has been found that pathological forms of TDP-43 or FUS can seed cytotoxic misfolding of endogenous wild-type superoxide dismutase in a prion-like fashion[18]. These inclusion bodies induce an immune-inflammatory reaction providing further damage to mitochondria and other cellular processes. The fact that ubiquitin positive cellular inclusions an impaired Ubiquitin-Proteasome System[20].
In this paper, we make use of a deep and broad search of the research literature to find papers related to four distinct topics: (1) The disease mechanism of ALS, (2) The metabolic pathways involved in the synthesis of glycosaminoglycans in the extracellular matrix and in glycoproteins, (3) epidemiological evidence of glyphosate as a causal factor in ALS as well as papers related to glyphosate’s mechanisms of toxicity, and (4) the many roles of glycine in proteins related to all of the above. We were motivated initially by the new paper by Wu et al.[6] showing the early involvement of gut dysbiosis in the disease process in a mouse model of ALS.
Our initial step was to enumerate most of the proteins which have genetic variants linked to ALS, and to specifically look for substitutions for glycine or mutations within glycine rich regions in these proteins in association with ALS. We were particularly struck by the association of fucose-depleted immunoglobulins with ALS, and this led to a thorough search for papers detailing all the enzymes and metabolic pathways involved in fucose synthesis, transport, and attachment to glycoproteins. We also explored the role of mineral imbalances in ALS, and the complex metabolic pathways related to fructose, a precursor to fucose. The novelty of our methods is the integration of research literature from diverse domains into a common story line, with particular emphasis on the presymptomatic stages of ALS.
In the remainder of this paper, we will fill in the gaps in the story of how ALS develops, showing how gut dysbiosis can eventually lead to motor neuron deterioration, with several intervening steps. Beginning with the disruption of the gut microbiome well in advance of overt symptoms of ALS, the pathology extends to metabolic disorders, particularly impairments in fructose metabolism, followed by steatosis, damage to skeletal muscle cells, and finally, the deterioration of the motor neurons in the spinal column. We will describe this entire cascade in the context of a theory that glyphosate, the active ingredient in the pervasive herbicide, Roundup®, can cause many of the impairments that are observed in association with ALS. Much of this disruption can be attributed to glyphosate’s ability to substitute for glycine during protein synthesis, acting as a glycine analogue[21]. Glyphosate’s disruption of mineral homeostasis likely also plays a role, particularly for manganese, copper and zinc[22-25].
Glyphosate acting as a Glycine Analogue
Glyphosate formulations are used extensively to control weeds growing among core crops in the processed food industry, particularly crops such as corn, soy, canola and sugar beets that are genetically engineered to be glyphosate tolerant. In the United States, usage of glyphosate has increased dramatically (100-fold) over the past two decades, in step with the wide spread appearance of glyphosate resistance among weeds[26,27]. While glyphosate is generally believed to be nearly nontoxic to humans, the World Health Organization’s IARC labelled glyphosate as a “probable carcinogen” in 2015[28]. Reliable tests have confirmed that humans are exposed to glyphosate. Krüger et al. measured levels of glyphosate in urine samples from several hundred humans, and found statistically significantly higher levels in those who were consuming a conventional diet compared to those consuming predominantly organic food (p < 0.0002) [29]. Furthermore, chronically ill people had significantly higher levels than people who were healthy (p < 0.03).
Recent evidence suggests that glyphosate has insidious toxic effects that are mediated in part by an impaired microbiome[27,30]. The key disrupted pathway in plants, the shikimate pathway, is also found in many of the gut microbes, and this pathway supplies essential aromatic amino acids and other nutrients to the host. Blockage in this pathway leads to an imbalance in gut microbes and disruption of fructose metabolism. As we will show here, this predicts profound effects on human physiology.
A more disturbing feature of glyphosate stems from its serving as an amino acid analogue of glycine. In Samsel and Seneff[21], it was proposed that glyphosate is able to substitute for glycine during protein synthesis by mistake. In fact, this is probably the way it works to disrupt EPSPS in the shikimate pathway. A strong case was made based in part on evidence of a precedent with other natural amino acid analogues that are produced by organisms during stress conditions. In fact, one of these, the non-coding amino acid β-methylamino-L-alanine (BMAA), produced by cyanobacteria, incorporates into proteins in place of L-serine, causing an ALS-like condition[31,32]. An epidemic of ALS in Guam is attributed to BMAA contamination in the seeds of cycad trees[31] and an extremely high rate of ALS in the Kii Peninsula, Japan, appears to have a similar etiology[32]. BMAA synthesized by cyanobacteria residing in the intestinal microflora has been implicated in ALS and Parkinson’s-Dementia complex as well as in equine motor neuron disease[33].
The rhizosphere is a term used to describe that area of soil involving the roots of plants and most notably the absorption of nutrients[34]. Interestingly, a study examining changes in bacterial gene expression in the rhizosphere following glyphosate treatment showed striking upregulation in proteins involved in both protein biosynthesis and protein degradation[35]. This suggests that glyphosate caused protein misfolding and induced energy wasting through repeated cycles of synthesis and disassembly of misfolded proteins.
Swanson et al.[36] have shown that many debilitating diseases are increasing alarmingly in the United States, in step with the dramatic rise in glyphosate usage on core crops. In Samsel and Seneff[21], it was shown how many of the diseases identified in the Swanson et al. paper can be explained through glyphosate substitution for glycine in specific proteins where glycine is highly conserved and plays an important functional role.
We hypothesize that the pathology leading to ALS begins slowly, but accelerates once liver function and the gut epithelial barrier have become severely compromised. Impaired gut barrier function allows glyphosate to reach the general circulation. Excess fructose may accumulate owing to impairments in fructose metabolism in the gut. The liver’s inability to clear fructose then stresses muscle cells, particularly under conditions of high physical activity and minimal adipose tissue. High protein turnover and accumulation of glyphosate embedded proteins in the muscles that cannot be cleared by the proteasome eventually leads to autoimmune reactions to proteins involved in muscle signaling. This in turn stresses the motor neurons that synapse with the diseased skeletal muscles. A die-back beginning from axon terminals eventually leads to cellular apoptosis of the controlling motor neuron, often associated with further development of autoimmune targeting of motor neuron proteins.
Evidence of a Link between Pesticides and ALS
While we suspect that the food is the most important source of glyphosate exposure in the general population, an agricultural worker is at much greater risk to occupational glyphosate exposure. While it is hard to obtain specific data on glyphosate, both pesticide exposure (OR = 1.44) and farming occupation (OR = 1.42) showed increased risk to ALS in a Korean study from 2014[37]. A survey in Michigan of 150 patients diagnosed with ALS found a much stronger link. Cumulative pesticide exposure was highly significantly associated with ALS (OR = 5.09, p = 0.002)[38]. It is likely that glyphosate was used at much higher levels in Michigan compared to Korea due to the heavy production of genetically engineered Roundup Ready soy in Michigan. Soybeans were Michigan’s largest export commodity in 2012, valued at over $800 million. A study based in Australia found an OR of 5.58 for exposure to industrial herbicides and pesticides, with a dose-response relationship[39]. A study based in Brittany obtained an odds ratio of 2.919 (p = 0.01) for an association between “agricultural activity” and ALS, and bulbar forms of the disease prevailed among those involved in agriculture[40].
Glyphosate has recently been shown to pass easily across epithelial mucosal barriers in the nasal cavity, via active transport by L-type amino acid transporters[41]. These authors wrote, “This additional pathway for glyphosate to enter the brain may result in much higher brain concentrations than previously anticipated based on oral or intravenous exposures, and may also explain the occurrence of reported neurologic toxicities.”
Resistance exercise increases the expression of amino acid transporters in muscle cells, including L-type amino acids (p < 0.05)[42]. This is expected as exercise increases muscle turnover and therefore requires protein synthesis. But this implies that a person who is athletic and physically fit is more likely to accumulate glyphosate in the muscle cells via transport through the L-type amino acid transporters, and it might help explain the observed increased risk to ALS among the physically fit[43,44]. Glyphosate residues have been detected in the muscles of chickens[45] and cows[29].
Table 1: Several proteins whose dysfunction is implicated in Amyotrophic Lateral Sclerosis and which have highly conserved essential glycines.
| Protein | Glycine Residue(s) | Reference(s) |
| SOD1 | G10, G37, G85, G93 | C Ricci et al. 2010[9] L Bruijn et al. 1996[8] |
| TDP-43 | C-terminal glycine-rich region | GS Pesiridis et al. 2009[63] |
| FUS | glycine-rich region critical for RNA binding | SK Dhar et al. 2014[61] |
| SLC351 | G180, G198, G277 | Zhang et al. 2012[80] |
| Complex I | Gx(x)GxxG motif | ME Baker et al.[66] |
| CcO | G283 | L Salomonsson et al. 2004 [87] |
| ubiquitin | C-terminal double-glycine | A Zuin et al. 2014[46] |
| myosin | G699 | NM. Kinose et al.,1996[175] |
| kinesin | G292 | BJ Grant et al., 2007[176] |
Roles of Conserved Glycines in ALS related Proteins
We noted previously that multiple glycine substitutions within SOD1 can produce an ALS mouse model. In fact, we have identified several proteins, in addition to SOD1, with highly conserved glycines that are implicated in the pathology of ALS, as shown in Table 1. Most intriguing is the fact that ubiquitin itself depends critically on a highly conserved carboxy terminal double-glycine pair to build the complex ubiquitin chains that signal a protein for degradation[46]. Substitution of glyphosate for either of these essential glycines would be expected to impair the process of recycling misfolded proteins. This could readily explain the accumulation of misfolded proteins that is a hallmark feature of ALS.
Protein aggregation of multiple proteins has been linked to ALS, including SOD1, TDP-43 and FUS. All three of these contain highly conserved glycines, and many of the ALS linked mutations are focused in glycine-rich regions[47,48], and often involve substitutions of other amino acids for highly conserved glycines. This implies that these glycines are protective against ALS, possibly through protection from protein misfolding.
Acylphosphatase is a small enzyme in which a large fraction of glycine residues are highly conserved across three domains of life[49]. An experiment involving systematically substituting six different glycine residues in human muscle acylphosphatase with other amino acids revealed that only G15A substitution causes a dramatic reduction in enzyme activity.
However, all other substitutions tested resulted in a marked increase in the protein’s tendency to aggregate. The authors concluded that the likely reason why these other glycines were highly conserved was to protect from protein aggregation. Glycine residues, due to the lack of a side chain, can occupy wider regions of conformational space. This leads to a substantial entropic penalty when a glycine residue converts from a disordered structure into a β strand, an important step towards aggregation. Substitution of glyphosate for any of these glycines can be predicted to also promote protein aggregation.
A seminal paper analyzed SOD1 for 150 missense mutations, and showed that, overall, the mutations led to a highly significant decrease in net charge and an increased tendency to aggregate[50]. The degree of change in stability and net charge were inversely correlated with ALS patient survival times. The author uses the term “proteome exhaustion” to describe his proposed theory for the underlying pathology linked to disease. The requirement for a high turnover rate to replace misfolded SOD1 molecules places a high energy demand on an otherwise already high-energy demand cell type: the motor neuron. This author wrote: “This analysis shows that protein misfolding diseases such as ALS are not necessarily caused by specific molecular toxicity of misfolded protein species, but possibly by systemic exhaustion due to elevated protein turnover. This mechanism could explain why identification of such malicious protein states has so far been unsuccessful.” Glyphosate substitution for glycine in any of the conserved glycines in SOD1 would lead to both an increase in negative charge and an increased tendency to aggregate. This could explain how even wild type SOD1 can be linked to ALS pathology[51,52].
Intriguingly, both TDP-43 and FUS are members of a broad class of proteins known as “RNA binding proteins.”[53]. FUS is involved in activation of translation from RNA to protein synthesis[54]. TDP-43 maintains protein quality control, and it responds to protein folding stress and regulates the levels of misfolded proteins[55]. Optineurin is another RNA binding protein, and it too has been linked to ALS[56]. Impaired optineurin function leads to progressive demyelination and axonal degeneration through receptor-interacting kinase 1 (RIPK1)-dependent signaling. RIPK1 activation is also induced by mutant SOD1[56]. Oligodendrocytes in particular are targeted, and impaired optineurin leads to impaired supply of myelin to the myelin sheath by oligodendrocytes.
RNA binding proteins respond to stress by inducing protein aggregation together with messenger RNA into “stress granules” (SGs) in a normally reversible process[53]. SGs in the cytoplasm are highly involved with RNA metabolism and homeostasis[53,57-59]. They are believed to function by accumulating at a site of specific cellular stressors such as alterations in homeostasis and temperature, external sources such as exposure to infection and chemicals as well as to internal sources such as mitochondrial and oxidative stress. Once thought to serve a role in the pathological process, it is now recognized that SGs function as a physiological process in a protective role. RNA-binding proteins recruit target mRNAs and round them up into SGs, which are then disassembled once cellular stress subsides[60]. This may be a way to temporarily halt synthesis of selected proteins. However, evidence points to pathological ALS proteins having the ability to dramatically disrupt SG function, thereby perpetuating further neuronal loss. In ALS, and in many other neurological diseases, accumulation of stress granules as protein inclusion bodies eventually disrupts cellular function.
It is striking that RNA binding proteins aggregate specifically through their glycine rich domains[53,61]. There is a 10-glycine stretch from amino acid residue 222 to 231 in exon 6 of FUS, and multiple cases of fALS are associated with deletions of several of those glycines[62]. The glycine-rich C-terminal region of TDP-43 is particularly aggregation prone. As shown in Figure 1, multiple missense mutations in this glycine-rich region have been linked to ALS[63]. Both of these proteins can be expected to be upregulated in response to the misfolding of SOD1. Cytotoxic granule associated RNA binding protein (TIA-1) is another RNA binding protein with a glycine-rich region[53].
It appears that there is evolutionary pressure to replace G93 in SOD1 with an alternative amino acid. Provocatively, this could mean that environmental factors, such as the vulnerability of glycine to glyphosate disruption, can influence mutation rates. Thus far, six distinct amino acid substitutions have been seen: serine, valine, asparagine, alanine, cysteine and arginine[64]. Interestingly, a common mutation, G93S, has a more virulent phenotype in the offspring compared to the parents. Averaged over nine parent-offspring pairs, the mean age of onset was 64.4 years in the parents, compared to 44.8 years in the offspring. An increase in a synergistic environmental factor such as glyphosate could explain this observation.
The short-chain dehydrogenase/reductase (SDR) super family represents one of the largest protein super families known to date. These enzymes catalyse nicotinamide adenine dinucleotide (P)(H)-(NAD(P)(H)-) dependent reactions, and their substrates are diverse, including polyols, retinoids, steroids, fatty acid derivatives and xenobiotics[65]. There are at least 73 members of this family in the human genome. They share a highly conserved glycine-rich structural motif, Gx(x)GxxG, with the NADH:ubiquinone oxidative reductive subunit of Complex I[66,67]. A highly conserved GxGxxG motif is found in NAD(P) H:quinone oxidoreductases from a variety of species[68]. This motif seems fundamental because enzymes that are not descended from a common ancestor share the motif. Patients with ALS have been shown to have impaired Complex I activity in their lymphocytes[69].
The hexosamine biosynthetic pathway begins with fructose-6-phosphate and glutamine as substrates, and the end product is uridine diphosphate N-acetylglucosamine (UDP-GlcNAc) a nucleotide sugar that is a precursor to many other derivative sugars that are incorporated into the heparan sulfate and chondroitin sulfate chains in glycosylated proteins.
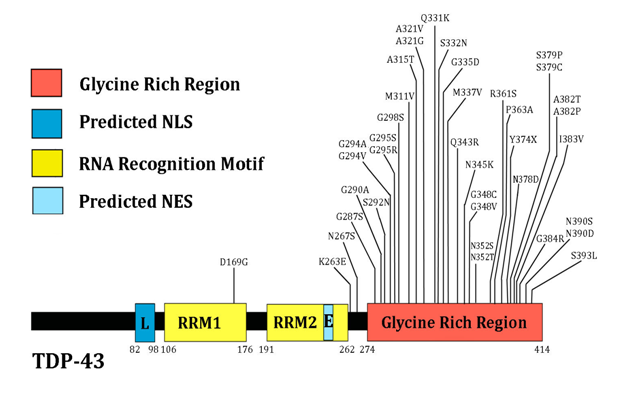
Figure 1: The multiple ALS-linked genetic variants of TDP-43 are concentrated in the C-terminal glycine-rich region. NLS: nuclear localization signal; NES: nuclear export signal; RRM: RNA recognition motif. Figure adapted from Lagier-Tourenne et al.[48]
Many of the enzymes that produce these derivative sugars, such as fucose and xylose, are NADPH-dependent members of the SDR family, and therefore they possess the Gx(x)GxxG motif that is essential for NADPH binding[70]. A G20A mutation in the GxxGxxG motif of FloA1, a UDP-GlcNac dehydratase in Helicobacter pylori, resulted in a complete loss of activity, suggesting that structural alterations of the nucleotide-binding site were involved[71]. A study on chondroitin sulfate chains in sea cucumbers showed that fucose branches induce resistance to degradation[72]. Deficiencies in these enzymes could lead to not only impairment in the ability to maintain the gut mucosal lining but also an accumulation of excess amounts of unmetabolized fructose-6-phosphate and glutamine.
More generally, the GxxG motif is important for stabilizing binding to both FAD and NAD(P) in oxidoreductases that are members of this broad but important class of enzymes[70]. Both UDP-GlcNAc and its derivatives, fucose and xylose, are essential constituents of glycosaminoglycans, proteoglycans, and glycolipids[73]. An important member of the SDR family is UDP-xylose synthase (UXS), also known as UDP-glucuronic acid (UDP-GlcA) decarboxylase and UDP-GlcA carboxylase, which catalyzes decarboxylation of UDP-glucuronic acid to produce UDP-xylose[74,75]. Xylosyl transferases are enzymes that catalyze the transfer of xylose from UDP-xylose to selected serine residues in the proteoglycan core protein[76]. This is the initial and rate-limiting step in mammalian glycosaminoglycans synthesis. Thus, a steady supply of UDP-xylose is absolutely essential for maintaining a healthy mucosal lining in the gut.
Not only are both xylose synthesis and xylose transport dependent upon essential glycine residues, but also the glyosylated protein’s attachment site for xylose is often glycine enriched. While the attachment site for the xylose sugar that initiates the synthesis of a chondroitin sulfate or heparan sulfate chain is a serine residue, this residue is always embedded in a short peptide sequence that is highly enriched in glycine residues[77]. The best acceptor protein known for xylosyl transferase is the alpha-trypsin inhibitor, bikunin. The chondroitin sulfate attachment site in this case includes a conserved glycine residue before the serine residue and a sequence of three glycine residues after the serine residue. A comparison of 50 different proteoglycans revealed a consistent pattern of a-a-a-a-Gly-Ser- Gly-a-Gly, where ‘a’ stands for any acidic amino acid. Thus, it can be expected that glyphosate substitution for any of these glycine residues would disrupt the attachment of xylose and therefore the initiation of synthesis of chondroitin sulfate or heparan sulfate. This is likely to be highly disruptive of the maintenance of the mucosal lining in the gut.
Transporter proteins involved in the synthesis of proteoglycans are also likely to be disrupted by glyphosate. There are several distinct transporters for the various nucleotide sugars involved in proteoglycan synthesis, and they belong to the protein class, SLC35[78,79]. Mammalian cells in the gut could be impaired in their ability to import fucose and xylose into the Golgi apparatus due to disruption of the glycines in these transporters through glyphosate exposure.
The GDP-fucose transporter SLC351 critically regulates the fucosylation of glycans[80], and defective versions are linked to serious disorders[78]. Three highly conserved glycine residues, Gly180, Gly198, and Gly277, in transmembrane helices 5, 6 and 8 of this transporter play essential roles in its activity. A substitution of tyrosine for Gly180 significantly diminished protein activity, and isoleucine substitution for Gly277 completely abolishedall cell surface fucosylation. It is interesting to note that glyphosate upregulates the expression (by at least a factor of 2) of four enzymes involved in fucose metabolism in E. coli: L-fuculose-1-phosphate aldolase, L-fucose isomerase, L-fuculokinase and fucose permease[81]. This may reflect a suppressed ability to incorporate fucose into branching glycans.
ALS patients have a deficiency in the serum levels of T-cell-expressed immunoglobulin Gs (IgGs), which are highly glycosylated proteins[82]. More specifically, the serum levels of fucosylated glycans in IgG N-glycans in ALS patients were found to be significantly reduced relative to the levels of sialylated glycans[83]. Under-fucosylated IgGs are also expressed in the motor cortex of ALS patients. A distinct fucose-deficient glycan, A2BG2, derived from ALS patient sera, enhances antibody- dependent cellular toxicity, and leads to over expression of CD16 and activated microglia in G93A-SOD1 mice[82]. Not only is A2BG2 specific for ALS, but also the amount produced is correlated with disease progression[84]. An active area of cancer drug research involves producing specific antibodies to proteins that tumor cells critically depend upon for survival. Special techniques have been developed to produce severely under-fucosylated antibodies in order to increase their toxicity to tumor cells[85]. Lack of core fucosylation in IgG results in a 50- to 100- fold increase in antibody-dependent cellular cytotoxicity[86]. It can therefore be predicted that under-fucosylated antibodies produced by ALS patients would be especially virulent in autoimmune disease.
Cytochrome c oxidase (CcO), also known as complex IV, is the last enzyme in the respiratory electron transport chain. Decreased CcO activity has been detected in the spinal cords of ALS patients[10,11]. CcO operates as both an enzyme catalyzing the reduction of oxygen to water and as a transmembrane proton pump. The segment that gates the protons overlaps the channel through which oxygen is delivered to the catalytic site. Replacement of a single glycine in a narrow part of this channel with valine completely blocks oxygen access to the catalytic site and results in the formation of a compartment around the site that is impermeable to small gas molecules. The catastrophic result is that it binds O2 several orders of magnitude more slowly than wild-type CcO[87]. This can be predicted to lead to leakage of superoxide and oxidative damage to neighboring vulnerable molecules.
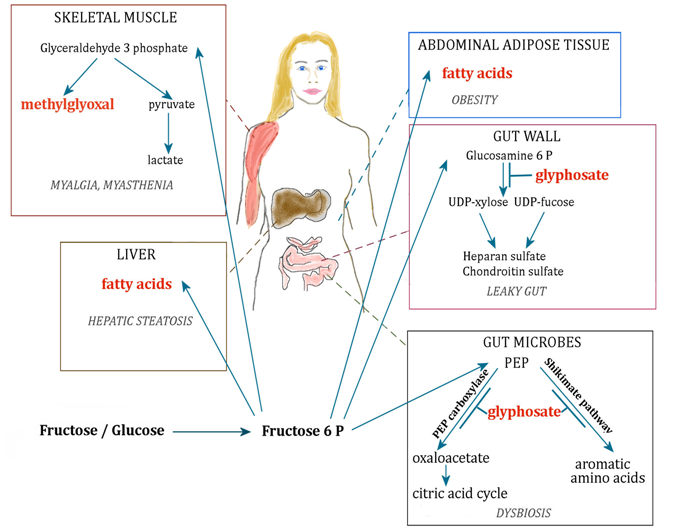
Figure 2: Schematic of metabolic pathways involving fructose and their disruption by glyphosate, leading to disease. PEP: phosphoenolpyruvate. Fructose 6 P: Fructose 6 phosphate. Glucosamine 6 P: glucosamine 6 phosphate.
Table 2: Amino acids paired with glycine in the dipeptide sequences synthesized from poly- GGGGCC, and their associated non-coding analogues. The CAG CAG repeat linked to Huntington’s disease is also included in the table, for completeness. Aze: Azetidine-2-carboxylic acid. DON: 6-Diazo-5-oxo-L-norleucine.
| Pattern | Paired Amino Acid | Analogue | Reference |
| GGG GCC | Gly-Ala | L-alanine | R S Roy et al.[212] |
| GGG CCG | Gly-Pro | Aze K | Bessonov et al.[213] |
| GGC CGG | Gly-Arg | L-canavanine | J Krakauer et al.[214] |
| CAG CAG | Gln-Gln | DON | CC Clark et al.[215] |
Metabolic Disturbances
Gut Dysbiosis
ALS is a debilitating disease with high morbidity and very poor treatment prognosis. Therefore, finding the early factors that eventually lead to the development of ALS seems like the best pathway towards conquering this disease. Thus, research efforts should be redirected towards uncovering the metabolic pathologies that precede overt expression of ALS symptoms. As schematized in Figure 2, we hypothesize that two important risk factors for ALS are excess dietary fructose from processed foods and chronic glyphosate exposure. As evidenced by the seminal paper on mutant SOD1 mice[6], the disease process likely begins in the gut. An imbalance in gut microbial distributions, particularly a deficiency in butyrate-producing species in the colon, leads to increased intestinal permeability and liver stress, likely due in part to impaired microbial clearance of fructose. The burden of fructose metabolism then falls primarily on the liver, which leads directly to hepatic steatosis. As the liver begins to fail, more fructose breaks past the hepatic barrier to enter the general circulation. As a consequence, skeletal muscles succumb to excess exposure to fructose and its highly glycating derivatives like methylglyoxal[88]. Over time, the skeletal muscles suffer increasing damage, in particular through concurrent exposure to glyphosate that puts an increased burden on the motor neurons to produce sufficient excitatory stimuli through neurotransmitter release. Eventually, the motor neurons suffer widespread damage and the overt symptoms of ALS become manifest. Mineral imbalances accelerate the decline.
The colonic environment is largely anaerobic, and the normal flora thrive by fermenting luminal complex carbohydrates derived from food sources or from the mucins produced by human goblet cells[89,90]. The colonic flora produce short chain fatty acids (SCFAs), including butyrate, succinate and propionate, and these are an important energy source for the colonic epithelial cells. Butyrate in particular has remarkable beneficial biological effects including histone deacetylase activity[91], and protection from colitis and colon cancer[90].
Butyrate producing bacteria are reduced long before overt ALS symptoms appear in an ALS mouse model[6]. N-acetyl tryptophan has shown therapeutic promise in a mouse model of ALS[92]. Tryptophan is a precursor for the quorum-sensing molecule indole, which mediates intercellular signaling among bacteria. Indole enhances barrier function of intestinal epithelial cells, by inducing the expression of genes responsible for tight junctions, adherens junctions, the actin cytoskeleton and mucin production[93,94]. In 2015, Parker proposed that glyphosate could induce impaired intestinal permeability in part through the reduction in supply of tryptophan as a consequence of its disruption of the shikimate pathway[95].
When the infant first adopts solid food, the intestine undergoes a profound transformation that involves a rapid growth of Bacteroides thetaiotaomicron, a species that is highly skilled in breaking down complex carbohydrates[96]. This species, in turn, induces the synthesis of fucosylated and sulfated glycans (mucins) by intestinal goblet cells. Bacteriodes species are adept at detaching these mucins from the intestinal wall and supplying them as a source of nutrition to other commensals, in addition to consuming them themselves. This symbiotic relationship between host and commensals leads to a healthy microbial distribution throughout life. Experiments with germ-free mice have shown that fucosylated glycan expression after weaning depends on the colonization by B. thetaiotaomicron[97]. B. thetaiotaomicron in particular is a dominating bacterium in the distal human gastrointestinal tract and plays a significant role to supply acetate to the butyrate-synthesizing microbes[98].
Bacteroides species are the most abundant gram-negative bacteria in the human gut, reaching a density of more than 1010 bacteria per gram of feces[99]. They have a tremendous capacity to degrade polysaccharides from both plants and the host organism. B.thetaiotaomicron, and, in fact, nearly all Bacteroides species, have two highly conserved and distinctly different forms of UDP-xylose synthase[100]. It has presented a puzzle to researchers as to why they synthesize UDP-xylose, because it isunclear what they use it for. Xylose is notably absent in bacterial cell walls, and they do not possess a xylosyltransferase enzyme.
We propose that their main purpose is to supply this important nutrient to the host to initiate glycosylation of membrane proteins so that the host can maintain a healthy mucosal environment for the bacterium. Significantly, mammalian cells secrete xylosyltransferase into the medium - the enzyme that attaches xylose to a serine residue in a glycoprotein. This fact also presented a puzzle to the researchers who investigated this, because the mammalian cell culture did not export any xylose to supply to the enzyme. We hypothesize that B. thetaiotaomicron and the host mucosal cells collaborate to produce the thick mucin walls of the colon.
An interesting recent study provides significant insight into how an important commensal microbe such as B. thetaiotaomicron is able to protect itself from immune attack induced by an inflammatory response to pathogens in the gut[101]. B. thetaiotaomicron resists binding to cationic antimicrobial peptides produced by the host immune cells through the activities of a lipid-A phosphatase enzyme that reduces the negative charge in the microbe’s outer wall by detaching phosphate groups from lipid-A. This phosphatase enzyme is a member of a broad class of lipid phosphatases that contain a highly conserved sequence of four peptides: PSGH[102]. While the significance of this peptide sequence to function is not yet clear, it can be predicted that substituting glyphosate for the glycine residue would likely disturb the enzyme’s behavior, and could lead to a reduction in B. thetaiotaomicron populations in the gut following an inflammatory response. Glyphosate has been patented as an antimicrobial agent, and it negatively impacts a large number of microbial species resident in the human gut[30]. The growth rate of Bacteroides in particular is severely reduced by glyphosate[103].
While dietary fiber has not been found to be protective against ALS[104], it must be kept in mind that important sources of fiber such as wheat, oats, barley, and legumes, are likely to be highly contaminated with glyphosate, due to the widespread practice of spraying these crops with glyphosate shortly before harvest as a desiccant[105]. Therefore, any potential benefits of the fiber may be offset by increased exposure to glyphosate.
Fructose Overload
Adipose tissue is a major metabolizer of fructose, probably exceeded only by the liver and gut[106]. Mouse fibroblasts in the presence of fructose as the only carbohydrate readily differentiate into adipocytes, suggesting that fructose is adipogenic[107]. A person with a lean body frame has very little adipose tissue, and therefore the ability to clear excess fructose through this pathway is severely restricted. Curiously, diabetes is protective against ALS, and this may follow from a reduced exposure of muscle cells to glycating sugars when insulin-based glucose uptake is impaired[108]. Anecdotally, an Italian professional soccer player who supplemented routinely with fructose 1,6 bisphosphate developed early onset of the bulbar form of ALS at the age of 45 years[109].
Glyphosate is frequently used as a ripener preharvest for sugar cane crops, and it increases the storage of sugars in the cane. It likely does this by impairing the synthesis of complex carbohydrates. A study on lupine plants showed that exposure to sublethal levels of glyphosate caused a decrease in starch content and an increase in sucrose[110]. The mechanism was attributed toinhibition of phosphoenol pyruvate carboxylase (PEPC), which is an important enzyme for the incorporation of both carbon and nitrogen into organic matter.
PEP is substrate to both PEPC and 5-enolpyruvylshikimic- 3-phosphate synthase (EPSPS), the enzyme that is disrupted in the shikimate pathway by glyphosate, leading to impaired synthesis of aromatic amino acids in plants and microbes. This is believed to be the main toxic effect of glyphosate to weeds. Both PEPC[111] and EPSPS[112,113] have essential glycines at the active site which, if replaced by glyphosate, would severely disrupt enzyme activity. This is probably the main explanation for its suppression of these enzymes. Changing the DNA code from glycine to alanine in E coli’s EPSPS completely abolishes glyphosate’s inhibiting effects[113]. Other microbes have acquired resistance through this same mechanism, and this is the basis for the bacterial gene modification in GMO Roundup-Readycrops[114].
Replacement of the terminal glycine in E. coli PEPC with a negatively charged amino acid such as aspartate resulted in a complete shutdown of enzyme activity[111]. These authors wrote: “PEPC appears to not tolerate additional negative charge at its extreme C terminus beyond that of the main chain free CO2 group.” Glyphosate substitution for the terminal glycine adds negative charge: it adds a CH2PO3H- anion at the C terminus. We hypothesize that the accumulation of PEP due to its blockage as substrate in these two important pathways would result in the inhibition of the pathway that converts fructose to PEP, leading to an accumulation of fructose and forcing more fructose to be phosphorylated to fructose 1,6 bisphosphate and fed into the glycolysis pathway. This likely also means that much of the dietary fructose is left unmetabolized by the gut microbes, instead placing a large burden on the liver to metabolize fructose, and explaining the epidemic we are seeing in fructose-induced fatty liver disease[115-117].
Sulfur Dysbiosis
Due to increased awareness of the benefits of butyrate to colonic health, a feeding study was conducted on rats to investigate the effect of different sources of protein and carbohydrate on butyrate production[118]. The rats were fed either fructooligosaccharide or a potato starch as carbohydrate, along with either casein or rice or soy as a protein source. An enhanced production of butyrate was associated with rice protein compared to casein or soy. We hypothesize that a factor in this observed difference was increased glyphosate contamination in the casein and soy, compared to rice. This is plausible given that most soy is engineered to be glyphosate resistant and casein is derived from cows that are fed high doses of glyphosate in their feed. Furthermore, they found that a poly-methionine supplement correctedthe low-butyrate yield for both casein and soy. A plausible hypothesis is that glyphosate interfered with methionine synthesis, but supplementation compensated for this. Methionine, a sulfur-containing amino acid, is essential for maintaining adequate levels of the important antioxidant glutathione in the liver, as well as the sulfonated amino acid, taurine, which is a significant component of bile acids.
A study on carrot cell lines showed that glyphosate reduced methionine levels by 50 to 65%[119]. A mechanism by which glyphosate could affect the synthesis of methionine by gut microbes is via glyphosate’s suppression of 3′-phosphoadenosine 5′-phosphosulfate (PAPS) reductase. A study on E. coli showed a 3.75 fold reduction in activity in the presence of glyphosate[120]. This enzyme is the rate-limiting enzyme in methionine synthesis. A structural analysis of the active site of yeast PAPS reductase rerevealed that it has three highly conserved motifs that collectively contain four glycine residues: two glycines in the 3′-P loop (51GLTG54), a single glycine in the middle of the so-called Arg loop, and a single glycine in the C-terminal catalytic motif, ECGIH, all of which are crucial to the protein’s enzymatic function[121].
A study on cows comparing a low-sulfur with a high-sulfur diet showed that sulfur was beneficial while the cows were grazing on grass, but that excess sulfur became problematic, causing a reduction in weight gain, once they were in the finishing stage[122]. Presumably, they were switched to a diet consisting of genetically engineered corn and soy feed that was likely heavily contaminated with glyphosate. Steers receiving the high-sulfur diet had a significant increase (p = 0.03) in sulfur reducing bacteria in the rumen, particularly Desulfovibrio desulfuricans, following adoption of the finishing diet. The high-sulfur diet resulted in a decrease in both butyrate and acetate compared to propionate, as well as an increase in the production of hydrogen sulfide gas. It is likely that glyphosate chelation of molybdenum contributes to this imbalance, as molybdenum catalyzes sulfite oxidase. Furthermore, copper suppresses the growth of Desulfovibrio species[123], so copper chelation by glyphosate might promote their growth. Strains of Desulfovibrio can utilize fructoseas an energy source to reduce sulfate to hydrogen sulfide gas[124].
Sulfur dioxide, sulfite and carageenan (sulfated polysaccharides extracted from seaweed) are commonly found in the modern Western diet[125]. High doses of these oxidized sulfur compounds, in conjunction with chronic glyphosate poisoning, can be expected to yield an overgrowth of sulfur-reducing bacteria in the colon at the expense of methanogenic species, and this has been directly linked to ulcerative colitis[125]. In vitro studies on isolated human colonocytes demonstrated that hydrogen sulfide specifically inhibits butyrate metabolism but not glucose oxidation by these cells[126]. H2S is implicated in ALS damage to motor neurons[127]. High levels of H2S have been found in the spinal fluid of ALS patients and in the tissues of mice with the SOD1G93A mutation[127].
One of the toxic effects of sulfite is to destroy thiamine, leading to a thiamine deficiency problem[128,129]. Thiamine deficiency has only recently been suspected as a causative factor in ALS. Following the deaths of two patients exhibiting signs of thiamine deficiency, researchers reporting in a 2015 paper investigated a potential role for thiamine deficiency in 122 patients with ALS[130]. They found severe thiamine deficiency in 18% of the patients with mild deficiency in another 10% of the patients. It may be highly significant that microbial thiamine synthase shares with ubiquitin the unique highly conserved double glycine repeat at the C-terminal end[46,131]. Remarkably, a paralytic disease that has afflicted multiple bird species in Northern Europe since the introduction of glyphosate in the early 1980’s has been clearly linked to thiamine deficiency[132].
Liver Disease and Fructose Metabolism
Patients with ALS tend to have exceptionally low levels of urate in their serum, and urate levels are inversely correlated with disease severity[133]. Urate is synthesized in the liver through degradation of purines in parallel with triglyceride synthesis. The rate-limiting enzyme, xanthine oxidase, depends on molybdenum as a cofactor. We have seen earlier that molybdenum deficiency could account for the toxicity of sulfite in the gut due to impaired sulfite oxidase activity. Defective xanthine oxidase could lead to an inability of the liver to metabolize fructose to fats, along with a decreased synthesis of urate.
Surprisingly, several studies have shown that alcohol consumption is protective against ALS. A meta-analysis involving 431,943 participants obtained an odds ratio of 0.57 for the association between alcohol consumption and ALS[134]. The mechanism remains unclear. This protection is not without cost, however, because alcohol severely stresses the liver, leading to steatohepatitis, liver fibrosis, and hepatic cancer. Interestingly, both fructose and alcohol increase both urate synthesis and triglyceride production in the liver, leading to nonalcoholic and alcoholic fatty liver disease, respectively[135]. However, they have opposite effects on the ratio of NAD+ to NADH, with fructose increasing it and alcohol decreasing it. Tryptophan (a product of the shikimate pathway) is an important precursor to NADH in the liver, but tryptophan is also converted into kynurenine and stored in macrophages invading an inflamed gut[136]. Both the suppression of the shikimate pathway and the induction of an inflammatory gut by glyphosate should therefore lead to an impaired supply of NADH to the liver, as discussed in Samsel and Seneff[30]. It is plausible that insufficient liver NADH prevents the liver from metabolizing fructose, but alcohol partially corrects this problem by renewing the supply through reduction of NAD+ to NADH, while simultaneously promoting liver disease. There could also be a more direct benefit through the high niacin content in beer, as a precursor to NAD. Alcohol will also promote synthesis of urate, both directly and indirectly through its positive effect on fructose metabolism in the liver. Urate is neuroprotective in part through its induction of the nuclear factor (erythroid-derived 2)-like-2 (Nrf2) signaling pathway in astrocytes[137].
In addition to molybdenum deficiency, NADH deficiency and an imbalance in the ratio of NAD+ to NADH, over activity of Desulfovibrio species in the colon could also contribute to impaired liver metabolism of fructose and other sugars. H2S is highly diffusible and can easily migrates from a source location to neighboring tissues. High levels of endogenous H2S produced in the gastrointestinal tract[138] would readily diffuse to the liver. It has been shown experimentally that the liver can consume large quantities of H2S, decreasing the NAD+/NADH ratio but consuming much of the available oxygen[139]. It can be predicted, therefore, that high exposure of the liver to H2S will compromise its ability to utilize oxidative phosphorylation to dispose of sugars arriving from the gut via the hepatic portal vein. In the case of a high fructose diet and poor fructose metabolism in the gut, this will pass the burden of fructose disposal mainly on to the skeletal muscle cells.
Even if the liver can clear much of the fructose in the presymptomatic phase of ALS, it is all the while accumulating excessive fatty deposits and suffering from cumulative damage due to chronic fructose exposure. In a rat model, copper deficiency enhanced the adverse effects of a high sucrose diet in the liver, leading to fatty liver disease, along with liver inflammation and fibrosis[140]. Eventually, liver disease will prevent the liver from further sustaining low serum levels of fructose. An investigation of liver function in a mutant SOD1G93A mouse model of ALS revealed a dramatic increase in the levels of natural killer T (NKT) cells in the liver, along with organ atrophy and the accumulation of stored fats. Even pre-symptomatic hepatic lymphocytes of these mice secreted significantly higher levels of cytokines when stimulated by an NKT ligand ex vivo[141].
Mineral Imbalances
Since both SOD1 and CcO are copper-dependent, it seems plausible that copper deficiency or excess could play a role in ALS. SOD1 is dependent on binding to both copper (Cu) and zinc (Zn) to function. Thus, an imbalance in the bioavailability of these essential minerals could lead to impairment. Remarkably, a strong case for both Cu deficiency[16,142-144] and Zn deficiency[145] as playing a pathological role in ALS has been made in the research literature.
CcO and Cu,Zn SOD (SOD1) are the two major copper-binding enzymes in humans. The copper chaperone for SOD, CCS, has a stronger affinity for Cu than the chaperones for CcO.In a double mutant mouse model involving both human SOD1 and human CCS (G93AxCCS mutant mice), CCS over-expression allows more Cu to be diverted into SOD, exacerbating any Cu deficiency problem induced by overabundant human SOD1 enzyme expression[142].
The Cu ATPase ATP7A is an important Cu-regulating protein which regulates both Cu(I) absorption from the small intestine and transport of Cu between the Golgi compartments and the plasma membrane of individual cells[146-148]. Mutations in ATP7A cause Menkes disease, a fatal infantile-onset neurodegenerative copper disorder, characterized by impaired billiary transport of Cu and many associated systemic pathologies[149,150]. ATP7A contains a glycine-glycine kink in the second transmembrane domain that forms a platform to accept Cu from the Cu-binding domains, to be pumped across the channel to the other side[149]. Glyphosate substitution for either of these glycines can be expected to disrupt this function.
A remarkable paper studying copper homeostasis in a mouse model of ALS proposed that the high copy number of human SOD1 in these mice places a high demand for Cu, resulting in general Cu deficiency[16]. They demonstrated that G93AxCCS mice developed ALS much more rapidly than single mutant SOD1 mice. These mice die about eight times faster than those without human CCS. Cu distribution is determined by affinity gradients, and SOD has the strongest affinity for Cu[151]. It is hypothesized that over-expression of CCS impairs Cu import into mitochondria, depriving CcO of Cu and thereby disrupting Complex IV[143,144]. Indeed CcO activity was greatly reduced in the SODG93AxCCS mice[143]. Remarkably, these double-mutant mice survived much longer if they were supplemented with a Cu complex, CuATSM. While this gives hope for a therapy for ALS patients, caution is necessary because excess Cu intake can cause toxicity.
Although we hear much from the research literature about the antioxidant tripeptide, glutathione, much less is written about a possibly equally important tripeptide, glycyl histidinyl-lysine (GHK), most often referred to as GHK-Cu due to its ability to carry Cu[152]. This Cu-chelating tripeptide plays an important role in delivering Cu to cells. Both of these tripeptides contain glycine, and one has to wonder about the consequences of glyphosate substitution for glycine in them. It can be predicted that a glyphosate-based version of GHK-Cu would bind Cu much more tightly, thus making it unavailable to SOD1and CcO.
A fascinating paper by Trumbull and Beckman discusses the role for Zn deficiency in the disease process[145]. Their compelling arguments show that Zn deficient wild type SOD1 is more destructive in inducing peroxynitrite synthesis from NO and O2. They further argue that Cu occupying the Zn site is much more redox active than Cu in the Cu site. They suggest that the benefits observed with Cu chelators such as d-penicillamine[153] and CuATSM[16] may be mainly due to their ability to extract Cu from the Zn site. Some of the mutant forms of SOD1 in familial ALS patients have a greatly reduced affinity to Zn (by as much as 30-fold)[154]. While Crow et al. proposed that this leads to enhanced tyrosine nitrosylation, another possibility is increased contamination of the Zn site by Cu.
Manganese (Mn) appears to be inappropriately distributed in association with ALS[155-157], generally showing up in excess. Manganism is a neurological condition known to be caused by excess Mn exposure among Mn smelters and miners and among welders[158], and it manifests as symptoms of both ALS and Parkinson’s disease. In a study by Kapaki et al.[155], Cu levels were found to be depleted in both the cerebrospinal fluid (CSF) and the serum in ALS patients compared to controls, whereas serum Mn levels were elevated. In Kapaki et al[156], a postmortem study on Mn levels in spinal cords showed significantly higher concentrations of Mn, particularly in the anterior horn. Kihira et al[157] found similar concentrations of Mn overall in the spinal column between ALS patients and controls, but there was an imbalance between the anterior horn and the posterior horn with excessive concentrations in the anterior horn in association with ALS. Roos et al[159] looked at Mn concentrations in the CSF and the blood, and found substantially higher Mn concentrations in the CSF in association with ALS (5.67 μg/L vs. 2.08 μg/L).
These odd distributions suggest that Mn may be distributed to the spinal column and CSF via a route that preferentially follows nerve fibers rather than through the circulation in ALS patients. In Samsel and Seneff[22], a link between abnormal Mn distribution channels and Parkinson’s disease was attributed to glyphosate’s disruption of bile flow through impairment of liver cytochrome P450 (CYP) enzymes. Normally, the liver redistributes Mn to the body through binding to bile acids. When this route is blocked, Mn is exported via the vagus nerve to first reach the brain stem nuclei and then spread from there, via nerve fibers, to other parts of the brain and spinal cord. Such pathways would explain both the high concentration in CSF and the excess Mn observed in the anterior horn, which is adjacent to the central canal in the spinal column. While we were unable to find any publications linking ALS directly to cholestasis, serum bilirubin










