(前回の続きである)
神経新生への影響
Impact on neurogenesis
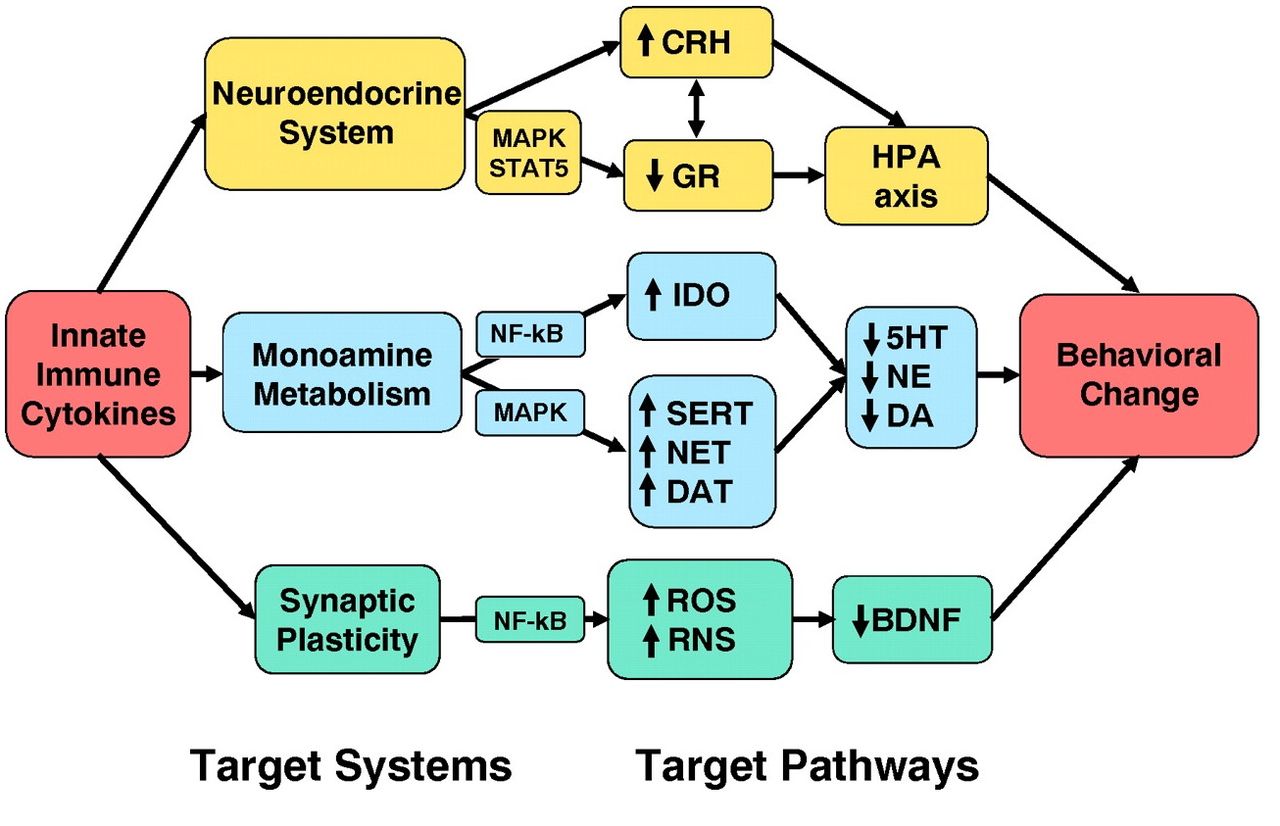
ある種の抗うつ剤が効果を発揮するためには神経新生が必要な要件となる。サイトカインは神経新生を阻害するというデータが示されている。従って、サイトカインは抗うつ薬の効果を阻害することができ、神経新生の抑制はサイトカインがTRDへと導く第2の主要な経路となる(図2B)。

炎症性サイトカインやサイトカイン誘導物質を投与すると、新たな神経細胞の増殖を減らし、これは特に海馬でで観察された。慢性的なストレスに晒された動物実験では、脳内におけるストレス誘発性の炎症性サイトカインの増加は、神経新生の減少とうつ病様行動に関連していることが示されている。ストレス状況下でも、炎症性サイトカインを阻害すれば、これらの効果を逆転できることが示されている。
IL-1受容体アンタゴニストを投与された動物や、IL-1受容体をノックアウトしたマウスでは、IL-1がブロックされているか、または、IL-1の効果が発揮されないため、神経発生や行動への慢性的なストレスの影響を逆にできることが示されている。神経新生をサポートする脳由来神経栄養因子(BDNF)などの成長因子への慢性ストレスによる抑制作用は、サイトカイン拮抗剤によって逆転される。インビトロやインビボ研究では、神経新生へのサイトカインの抑制効果はNF-κBの活性化により部分的に媒介されていることを示唆しており、海馬における神経幹細胞様細胞の増殖を直接阻害している。
なお、抗うつ剤の抗うつメカニズムの一部はサイトカイン(特に、炎症性サイトカイン)の誘導が関係していることも分かっており、必ずしもサイトカインを 抑えれば良いという訳ではない。SSRIのシタロプラムやNaSSAであるミルタザピンはIL-1β、TNF-αを増加させるこという報告もある。このあたりのサイトカインが絡む抗うつ効果のメカニズムはまだよく分かっていない。
なお、抗うつ剤の抗うつメカニズムの一部はサイトカイン(特に、炎症性サイトカイン)の誘導が関係していることも分かっており、必ずしもサイトカインを 抑えれば良いという訳ではない。SSRIのシタロプラムやNaSSAであるミルタザピンはIL-1β、TNF-αを増加させるこという報告もある。このあたりのサイトカインが絡む抗うつ効果のメカニズムはまだよく分かっていない。
グルタミン酸への影響
Impact on glutamate
神経の可塑性や興奮性神経伝達におけるグルタミン酸の重要性が考察され始め、感情障害におけるグルタミン酸の役割、特に、TRDにおけるグルタミン酸の役割への認識が高まってきている。グルタミン酸N-メチル-D-アスパラギン酸(NMDA)受容体拮抗薬であるケタミンがTRDを有する患者に劇的な治療反応を生じさせる事実は、グルタミン酸アンタゴニストにて大うつ病が治療できる可能性を示唆している。従来の抗うつ薬は、グルタミン酸受容体に何らかの効果を有しており、動物実験でもストレスによって誘発されるグルタミン酸の放出を阻害することが見出されたが、この従来の抗うつ薬のグルタミン酸への効果は、モノアミン神経伝達への効果を介した第一次的な効果なのか二次的な効果なのかは不明なままである。
炎症性メディエーターがグルタミン酸神経伝達の増加を引き起こすメカニズムとしては以下のものが考えられている。
(1) NMDA機能のアップレギュレーションと増強が生じる
(2) グルタミン酸の放出が増加し、グルタミン酸の再取り込みが阻害される
(3) AMPA受容体への作用
(4) キヌレニン経路の活性化
炎症性サイトカインは、アストロサイトやミクログリアといったグリアエレメントにおけるグルタミン酸の再取り込みとグルタミン酸トランスポーターの発現を減少させる。これらの効果を媒介するであろうと考えられている1つのメカニズムが示されているが、それは反応性窒素や酸素種(活性酸素、ROS)の誘導であり、ROSはアストロサイトにおけるグルタミン酸の取り込みに影響を与えることができる。

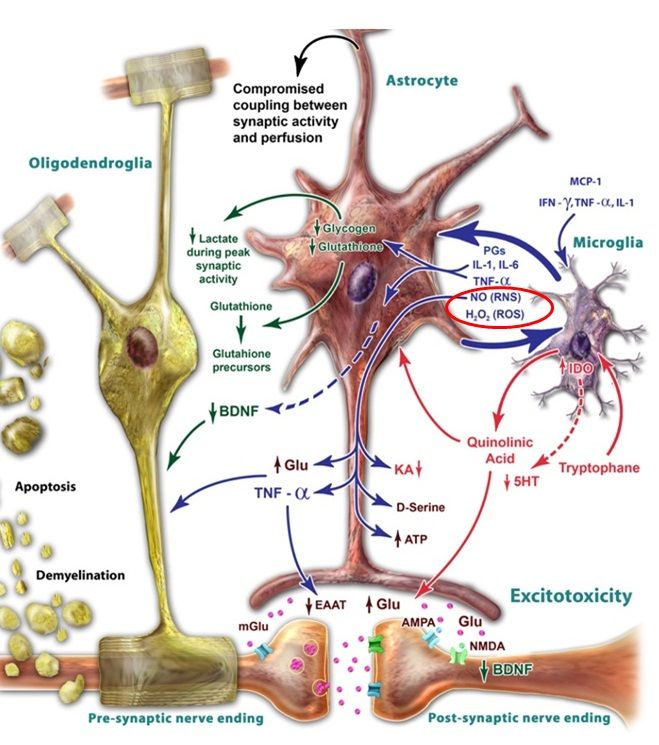
一酸化窒素による炎症性サイトカインの誘導は、アストロサイトからのグルタミン酸の放出を刺激するが、この効果は一酸化窒素合成酵素を阻害することで逆転できることが示されている。アストロサイトからのグルタミン酸の放出は、シナプス外のNMDA受容体へアクセスし、有害な結果をもたらす。すなわち、シナプスのNMDA受容体よりもシナプス外のNMDA受容体の活性化はBDNFの発現を減少させ、細胞死を強めることになる。
炎症性サイトカインによるIDO酵素の誘導もグルタミン酸興奮毒性に寄与する可能性がある。IL-1βやTNFαに暴露された海馬ニューロンではNMDA受容体やAMPA受容体を介して誘導される興奮毒性による神経損傷を強めることが示されている。IDOはトリプトファンからKYNへの変換を誘導するが、脳内の細胞に順次アクセスして、ミクログリアおよびマクロファージではさらにキノリン酸(quinolinic acid、QUIN)に変換されて、炎症プロセスの間に脳へ浸潤していく。QUINは直接NMDA受容体に結合するため、興奮毒性の活性化に寄与することになる。さらに、QUINはグルタミン酸の再取込みをも阻害し、アストロサイトからのグルタミン酸の放出をも刺激することが示されている。
さらに、TNFαは、AMPA型グルタミン酸受容体の輸送に影響を与えることが示されている。AMPA受容体は通常では4つのサブユニット(GluR1-4)を持っている。TNFαはGluR2のサブユニットを欠くAMPA受容体の産生をもたらす。この受容体の立体構造は、神経細胞へのカルシウム流入を促進する。このAMPA受容体の立体構造の変化は、グルタミン酸によるニューロンへの興奮毒性を誘導することになる。
一方、QUINは酸化ストレスの強力な誘導物質でもある。うつ病に関連する所見として、インターフェロンαで治療されたうつ病患者の脳脊髄液中のQUIN濃度の上昇が報告されており、この所見はうつ病と炎症性マーカーとも相関していた。うつ病患者の死後脳の調査では、ミクログリアにおけるQUIN免疫反応性の増加が前帯状皮質(ACC)の膝下(subgenual)と中帯状(midcingulate)領域で見出されている。
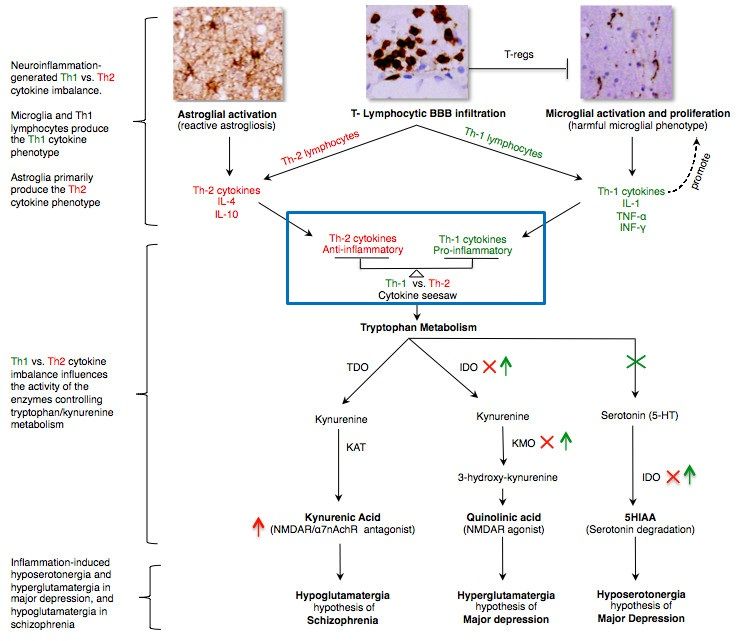
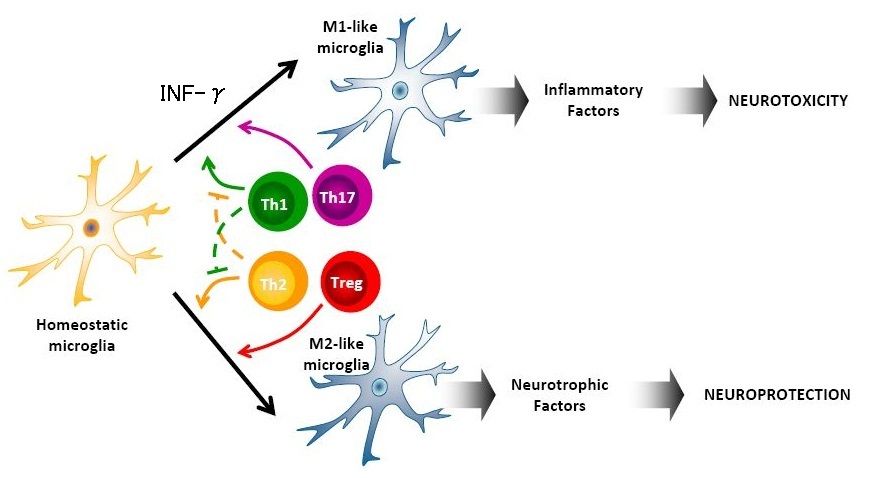
PMC3626880によれば、大うつ病や統合失調症でのトリプトファン・キヌレニン系の代謝不全はグリア細胞やTリンパ球によって生成される「Th1とTh2からのサイトカインによるシーソー」として理解すべきだと述べられている(下図)。大うつ病ではTh1からのサイトカインが優位になっていると考えられている(逆に、統合失調症ではTh2のサイトカインが優位になっているとPMC3626880の著者は仮説を立てている)。

大うつ病では、末梢の炎症により末梢のTH-0リンパ球が活性化されBBBを通過しCNSに侵入するようになる。CNSに侵入したTH-0リンパ球は、本来は神経保護的に作用するはずのミクログリアの過剰なTh1応答を引き起こしてしまい、その結果、CNSのグルタミン酸過剰が惹起され、神経興奮毒性の方向への変化に傾いてしまっているのだろうと考えられている(下図)。
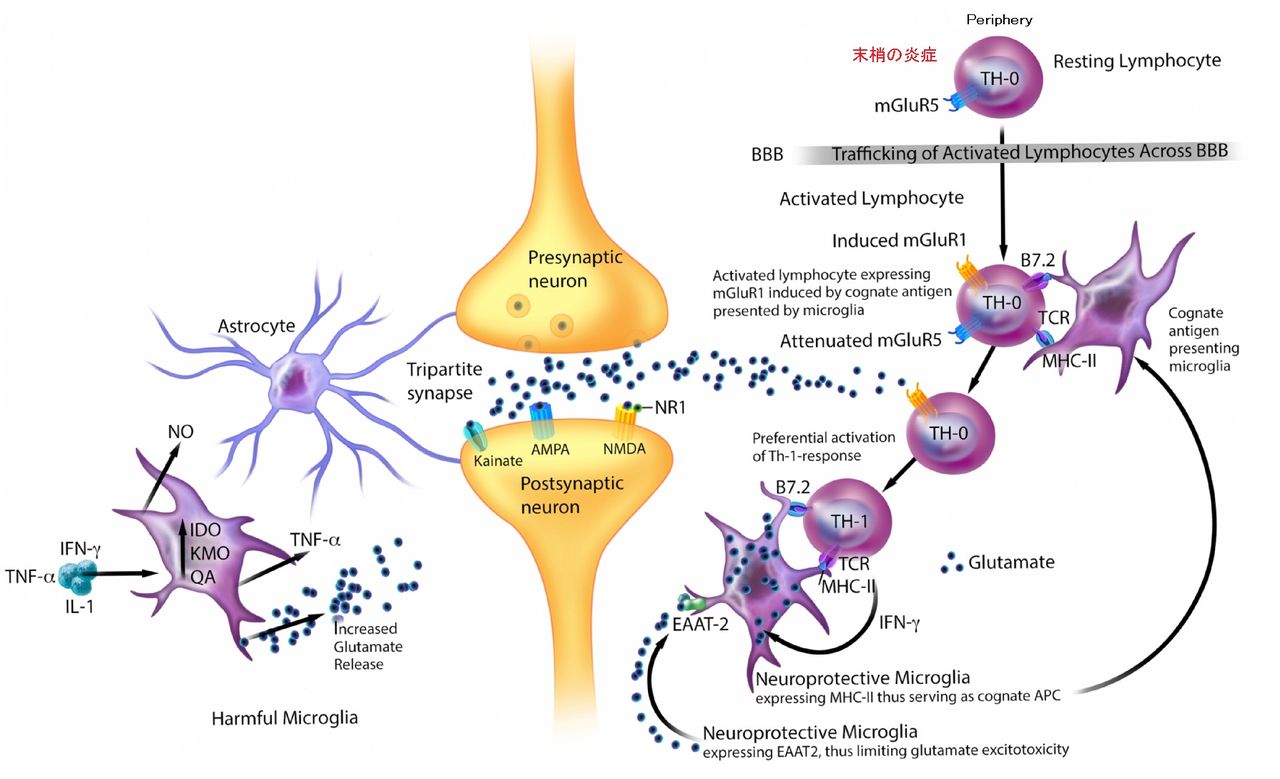
一方、グルタミン酸の過剰そのものがTh1の反応を強めてしまう。すなわち、Th1⇔グルタミン酸との間で悪循環に陥っている可能性があることになる。動物実験では、リンパ球のmGluR1受容体への過剰なグルタミン酸の結合は、IFN-γなどのTh1サイトカインの産生を促進してしまうことが分かっている。
大うつ病では皮質のグルタミン酸のレベルがうつ症状と相関していることが報告されている。サイトカインはアストロサイトだけでなくミクログリアにおいてもQUINの合成を増加させ、シナプスへのグルタミン酸の放出を促進させる。さらに、QUIN自体がアストロサイトのカルシウムの流入を増大させ、アポトーシスを促進させてしまう。こういったメカニズムによってグリアも障害されていくことになる。
グリアへの影響
Impact on gliaelements
このようにして、炎症によりグリアも障害されていく。大うつ病の死後脳の調査では、アストログリアの損失が前帯状皮質、前頭前野、扁桃体、白質などで見出されている。うつ病が長引くほどアストログリアも失われていくようである。ラットではアストログリアへ毒性を示す薬剤の投与によってうつ病様状態が引き起こされる。
障害をうけるのはアストロサイトだけではない。髄鞘(ミエリン)形成に大きく関与しているオリゴデンドログリアも影響を受けることが分かっている。大うつ病ではオリゴデンドログリアの減少が報告されている。従って、ミエリンを修復する目的でω3脂肪酸を補給することには大きな意味があろう。
さらに、炎症によってミクログリアにも変化が生じている。ミクログリアは神経保護の方向にも作用するが、神経を障害する方向にも作用する両刃の剣である。ミクログリアの活性化と増殖(microglial activation and proliferation、MAP)は神経損傷の指標となる。精神疾患におけるMAPに関する所見はまだ一定していないが、大うつ病などではMAPの上昇が報告されている。自殺したケースではキノリン酸陽性のミクログリアの細胞密度が膝下前帯状皮質で増加していた。
細胞外の過剰なグルタミン酸はTh1反応を強め、INF-γが誘導され、INF-γは少量であってもミクログリアの表現型を神経障害性(M1-like)へと変えてしまう。逆に、細胞外の過剰なグルタミン酸が除去されれば、神経障害性へと表現型が変化してしまったミクログリアも細胞保護性へと形質転換されるらしい。

本年度に発表された論文では、ストレス刺激(5週間)によって、最初の1週間以内にミクログリア活性化と増殖が生じたが、逆に、5週後には、海馬ではアポトーシスにてミクログリア数は減少し形態も痩せ細ったように変化し、うつ病様行動を示すようになったと報告している。うつ病様行動は、ストレス刺激の初期にミノサイクリン、IL-1受容体のブロック、イミプラミンで処置されることによって阻止された。一方、5週以降の時期では、ミノサイクリンやイミプラミンではうつ病様行動への効果がなかったが、ミクログリアの増殖を刺激する因子であるマクロファージコロニー刺激因子(macrophage colony-stimulating factor)や顆粒球マクロファージコロニー刺激因子(granulocyte-macrophage colony-stimulating factor)によって海馬での神経新生が促されうつ病様行動は逆転された。これらの知見はミクログリアの数が減少してしまった慢性のストレス状態では抗うつ剤の効果は期待できないことを意味する。
また、グリアの大きさも変化するようであり、大うつ病ではアストログリアのサイズの減少が報告されている。逆に、自殺をしたうつ病患者の死後脳からは前帯状皮質のアストログリアのサイズが増加している所見が見出されている。アストログリアのサイズに関する報告は一定はしていないようである。なお、双極性障害では、リチウムやバルプロ酸によって治療しコントロールができたケースではグリアの損失の大幅な改善を認めたという報告がある。
ミクログリアの活性化によって炎症性サイトカインや酸化窒素(NO)が誘導されるが、これらによって酸化ストレスが強まることになる。酸化ストレスによって、膜リン脂質などの膜成分が損傷し、細胞内の抗酸化物質(グルタチオンなど)は枯渇していき、脂質の過酸化を招く活性酸素種(ROS)が上昇していく。ROSはさらにNF-κBを刺激して、炎症誘発性物質の産生が誘導される。その結果、神経障害を招き、グルタミン酸の放出を増やし、グルタミン酸によってミクログリアがますます活性化される・・・・・。このようにROSを介した炎症・酸化ストレス(ROS)・グルタミン酸という悪循環のループが神経細胞⇔グリアエレメントの間で形成されていく。
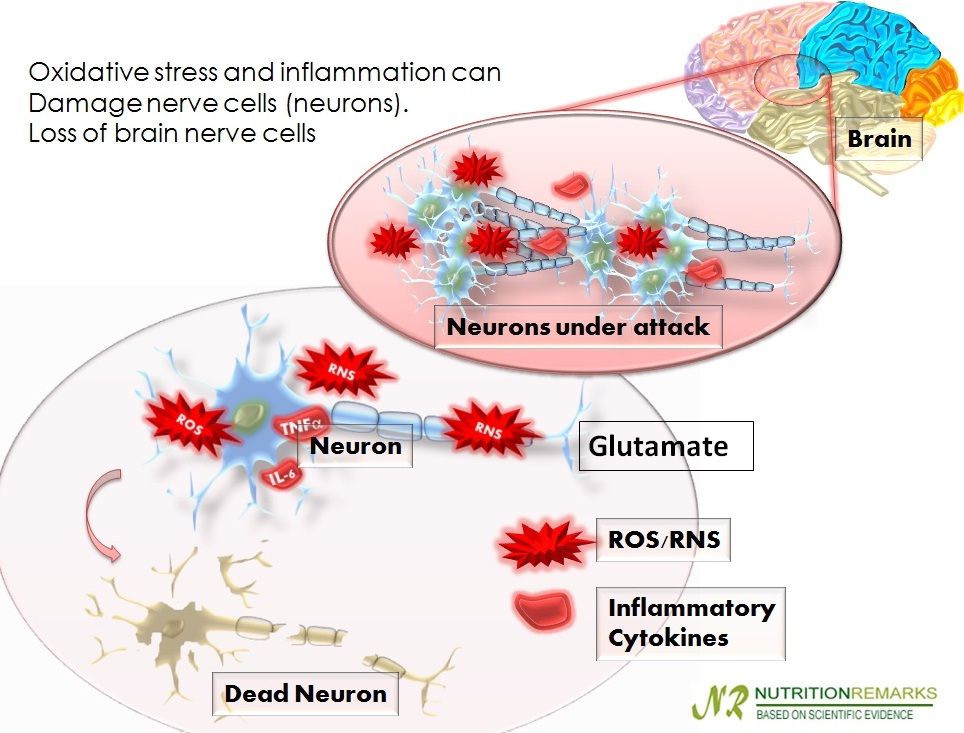
神経炎症と酸化ストレスの増大
Neuroinflammation and increased oxidative stress
酸化ストレスは酸化物質の過剰に陥っている状態であり、酸化ストレスによって、脂質、タンパク質、DNAなどの生体高分子が損傷を受ける。臓器の中でも特に脳は酸化ストレスに対して脆弱である。なぜならば、(1)脳は過酸化が可能な多価不飽和脂肪酸が多く含まれている。(2)脂質を過酸化反応させる酸素ラジカルを誘導する微量ミネラル(鉄や銅など)が多く含まれている。(3)酸素利用率が高い。 そのため、脳においては抗酸化メカニズムが発揮されることが限られてしまうためである。
大うつ病、双極性障害、統合失調症、強迫性障害などでは酸化ストレスが生じている可能性が高い。末梢性マーカーとして、脂質過酸化生成物(例えば、マロンジアルデヒドや4 -ヒドロキシ-2 -ノネナール)を含む脂質過酸化生成物質の増加、一酸化窒素(NO)代謝物の増加、抗酸化物質(例えば、グルタチオン)の低下と、抗酸化酵素のレベルの変化が報告されている。
大うつ病では、スーパーオキシド・アニオン・ラジカルの生成増加と、酸化を媒介する好中球によるアポトーシスが上昇している。抗酸化酵素の血清レベル(例えば、スーパーオキシドジスムターゼ-1)は、急性うつ病性エピソードでは上昇しているが、SSRIによる治療後に正常化されることが報告されている。大うつ病では、血清抗酸化酵素のレベルは酸化ストレスの増加による急性の効果を相殺する代償機構を反映しているのだろう。大うつ病とは対照的に、早期に発症した統合失調症では、CSFの可溶性スーパーオキシドジスムターゼ-1レベルは、実質的に減少している。これは脳の抗酸化酵素レベルの減少は酸化的損傷を生じさせ統合失調症の発症に関連することを示唆している。
うつ病の動物モデルでは、脳の脂質過酸化レベルやNOレベルが上昇している際には、グルタチオンの脳レベルも減少していることが分かっている。大うつ病、双極性障害、統合失調症の死後脳研究では、脳内の総グルタチオンのレベルの低下が報告されている。うつ病における酸化ストレスの主要なメカニズムとしてグルタチオンの枯渇が考えられているが、大うつ病の患者から培養した線維芽細胞では、グルタチオンのレベルには依存しない酸化ストレスの増加も示されている。
ミトコンドリアの機能障害も、大うつ病、双極性障害、統合失調症で増加しうる酸化ストレスに貢献する可能性がある。ミトコンドリア病における死後脳の研究では、ミトコンドリアDNAの異常が明らかされ、精神障害の有病率が高さと相関していた。インビトロの動物実験では、TNF-αなどの炎症性サイトカインは、ミトコンドリアの密度を低下させ、ミトコンドリアにおける酸化エネルギー代謝障害し、ROS産生の増加をもたらすが示唆されている。これらの実験結果は、神経炎症・ミトコンドリア機能障害・酸化的ストレスとの間をリンクさせるメカニズムが存在することを示唆している。
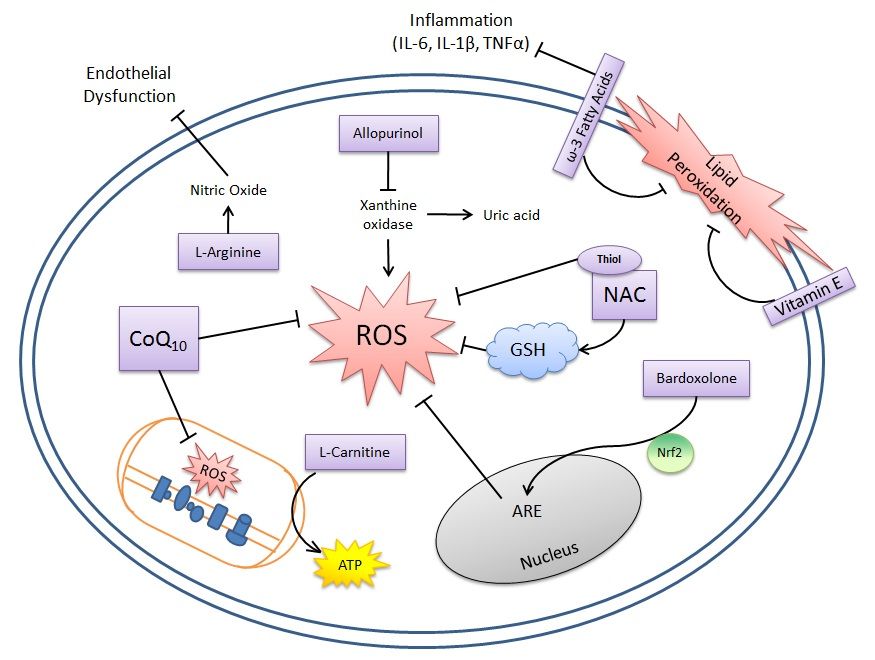
この炎症と酸化ストレスが絡む悪循環を断ち切るのがN-アセチルシステイン(NAC)であり、大うつ病、双極性障害などで有効性がRCTにて既に実証されている。抗酸化物質であるグルタチオンを補給することも炎症と酸化ストレスの悪循環を断ち切る上では意味がある方法となろう。グルタチオンは経口で投与しても神経系には届かないようであり、非経口で投与する必要がある。さらに、抗精神病薬、SSRI、気分安定剤は抗酸化特性を有することが示唆されている。なお、NAC以外の抗酸化物質(例えば、ビタミンCとEなど)のアジュバントとしての治療的役割はRCTが行われたが実証はされなかった。ROSによる悪循環を断ち切る可能性がある物質を下図に示す。

血液脳関門の機能障害
Blood brain barrier dysfunction
一方、脳血液関門(BBB)も炎症によって障害されていく。以前は、脳はBBBによって保護されており、免疫特権状態にあると考えらてきた。しかし、現在では、脳はBBBによって必ずしも守らている訳ではないことが分かっている。大うつ病や統合失調症などではCSF:血漿のアルブミン値の上昇からBBBの透過性が亢進していることが推測されている。さらに、大うつ病などの死後脳の超微細構造を調べた病理組織所見でもBBBが障害されている所見が得られている。
活性化したミクログリアは炎症性サイトカインを増大させ、NOを増加させ、PGE2の産生を介してBBBの血管内皮細胞におけるCOX2を増加させる。そしてCOX2によってBBBの血管内皮細胞が障害されていくことになる。それによってBBBの透過性が高まることになる。BBBの障害が大うつ病や統合失調症で報告されている以上、COX2阻害剤の投与は炎症からBBBを守る上では意味のある方法だと思われる。さらに、前回のブログで述べたように、L-methylfolateや葉酸による補充療法はBH4を介したBBBの保護作用が期待できる方法となろう。

S100Bの役割
The role of S100B
サイトカインはアストログリアからのS100Bの分泌も増大させる。S100Bはアストロサイトやオリゴデンドログリアによって産生される分子量が10 kDaのカルシウムと結合するタンパク質である。S100Bは濃度によって作用が異なってくる。ナノモルレベルではストレスによる神経損傷を抑制し、ミクログリアからのTNF-αの放出を阻害し、アストログリアにおけるグルタミン酸の取り込みを増加させる。すなわち、神経保護の方向に作用する)。しかし、マイクロモルのレベルにまで濃度が上昇してしまうと、神経細胞のアポトーシスを誘導し、COX2/PGE2やIL-1βの産生を増加させ、NOを誘導し、モノサイトやミクログリアからのTNF-αの放出を高めてしまうため神経を障害する方向に作用してしまう。
脳脊髄液(CSF)や脳組織中のS100Bはアストログリアの活性化の指標となる。大うつ病では血清中のS100Bの濃度と自殺傾向との相関が示されている。急性うつ病エピソードや躁病エピソードにおける血清やCSFにおける上昇が報告されている。なお、統合失調症の死後脳においても上昇が報告されている(ACC、dlPFC、OPCなどの領域)。S100Bの上昇は、妄想、陰性症状、認知障害、治療反応性の低下などと関連している。
(次回に続く)
このブログにコメントするにはログインが必要です。
さんログアウト
この記事には許可ユーザしかコメントができません。